Peptic Ulcer Disease
Peptic ulcer disease is a condition characterized by mucosal defects in the stomach or duodenum that extend through the muscularis mucosae, resulting from an imbalance between gastric acid–pepsin aggression and mucosal defense mechanisms.
Peptic Ulcer Disease (PUD) — Definition, Epidemiology, Risk Factors, Anatomy, Etiology, Pathophysiology, Classification & Clinical Features
Peptic ulcer disease (PUD) refers to defects in the gastrointestinal mucosa that extend through the muscularis mucosae into the submucosa or deeper layers, occurring in areas exposed to peptic (acid-pepsin) activity [2]. The key distinction from an "erosion" is depth: an erosion is superficial and does not breach the muscularis mucosae, whereas an ulcer does — this is why ulcers bleed from submucosal vessels and can perforate.
Breaking down the name:
- "Peptic" → from Greek peptikos ("able to digest") — these ulcers arise in areas bathed by gastric acid and pepsin.
- "Ulcer" → from Latin ulcus ("a sore") — a full-thickness mucosal defect.
So the name literally means "a digestive sore" — a mucosal defect caused by acid-pepsin digestion overwhelming mucosal defence.
Core Concept — Ulcer vs Erosion
An erosion is confined to the mucosa (does not breach muscularis mucosae) and heals without scarring. An ulcer penetrates through the muscularis mucosae into the submucosa — this is why ulcers can erode into arteries (e.g. gastroduodenal artery), cause significant haemorrhage, perforate, and heal with fibrotic scarring (which can cause strictures / gastric outlet obstruction).
2. Epidemiology
- Lifetime prevalence: ~5–10% of the population will develop a peptic ulcer [2].
- Male predominance (M:F ≈ 3:1), although the gap is narrowing with ageing populations and increased NSAID use in women [2].
- Duodenal ulcers (DU) account for ~75% of peptic ulcers; gastric ulcers (GU) ~20%; the remainder occur at other sites (oesophagus, Meckel's diverticulum, anastomotic sites) [2].
- In Hong Kong, PUD remains the most common cause of upper GI bleeding [1][3], largely driven by:
- High prevalence of H. pylori infection (declining but still significant in older cohorts — historically > 50% seroprevalence in adults > 60 years).
- Widespread NSAID and aspirin use in an ageing population with cardiovascular and musculoskeletal disease.
- Stress-related mucosal disease in ICU patients.
- PUD is the leading cause of death among peptic ulcer complications — specifically haemorrhage [3].
High Yield: Peptic ulcer is the most common cause of UGIB [1]. The four major risk factors are H. pylori, NSAIDs, stress, and excess gastric acid [1][3].
- Overall incidence of PUD has declined due to H. pylori eradication and PPI use.
- However, NSAID/aspirin-related ulcers are increasing relative to H. pylori ulcers as eradication rates rise but antiplatelet/anticoagulant use expands.
- The proportion of "idiopathic" ulcers (neither H. pylori nor NSAID) is rising — now ~10–20% of DU and ~5–10% of GU in some series.
3. Anatomy & Function
The stomach is divided into five anatomical regions (proximal → distal):
| Region | Key Features |
|---|---|
| Cardia | Surrounds the gastro-oesophageal junction (GOJ); mucus-secreting glands |
| Fundus | Superior dome; contains many parietal (oxyntic) and chief cells |
| Body (Corpus) | Main acid- and pepsinogen-secreting region; parietal cells here are the target of vagotomy |
| Antrum | Produces gastrin (G cells) and contains mucous glands; also has D cells (somatostatin) |
| Pylorus | Sphincteric muscle controlling gastric emptying into the duodenum |
The duodenum is divided into four parts (D1–D4). D1 (the duodenal bulb/cap) is the most common site for duodenal ulcers because it receives the highest concentration of gastric acid exiting the pylorus before alkaline pancreatic/biliary secretions neutralise it in D2 [2].
Why D1?
Acid load is maximal in D1 because pancreatic bicarbonate enters via the ampulla of Vater in D2. D1 therefore has the highest acid exposure and is where DU almost always occur. An ulcer beyond D1 (e.g. D2, jejunum) should raise suspicion for Zollinger-Ellison syndrome or other unusual aetiologies [2].
Understanding the arterial anatomy is critical because ulcer haemorrhage occurs when the ulcer erodes into an artery.
Greater curvature:
- Short gastric arteries (from splenic artery)
- Left gastro-omental (gastroepiploic) artery (from splenic artery)
- Right gastro-omental (gastroepiploic) artery (from gastroduodenal artery)
Lesser curvature:
- Left gastric artery (from coeliac trunk — the largest blood supply to the stomach)
- Right gastric artery (from common hepatic artery / proper hepatic artery)
Duodenum (D1):
- Supplied by branches of the gastroduodenal artery (GDA), which runs behind the first part of the duodenum.
Clinical Pearl — The GDA and Posterior DU Bleeding
Posterior duodenal ulcers classically erode into the gastroduodenal artery (GDA), causing massive, life-threatening haemorrhage (coffee-ground vomiting or melena) [3]. This is the single most tested vascular anatomy fact in PUD.
Anterior duodenal ulcers tend to perforate into the peritoneal cavity (because there is no adjacent artery to tamponade; the anterior wall faces the free peritoneal space).
Mnemonic: "Posterior bleeds, Anterior perforates" — or "Posterior Pours, Anterior Perforates."
Gastric ulcers on the lesser curvature can erode into the left gastric artery — the largest arterial supply to the stomach — causing catastrophic haemorrhage.
Sympathetic supply:
- Greater splanchnic nerve (T5–T9 sympathetic trunk) → coeliac ganglion → stomach
- Function: inhibits peristalsis, constricts pyloric sphincter, reduces secretions
Parasympathetic supply (Vagus nerve, CN X):
- Anterior vagal nerve: innervates the stomach, pylorus, and liver (gives off the hepatic branch, then continues as the anterior nerve of Latarjet along the lesser curvature) [2]
- Posterior vagal nerve: innervates the stomach and the entire foregut/midgut down to the splenic flexure (gives off the coeliac branch, then continues as the posterior nerve of Latarjet) [2]
The nerve of Latarjet (anterior and posterior) runs along the lesser curvature and provides the principal motor and secretory innervation to the antrum and pylorus. It is the key structure in highly selective vagotomy (HSV), where only the parietal cell branches are divided while preserving the nerve of Latarjet to maintain antral motility and pyloric function [3].
High Yield for surgery: In truncal vagotomy, both vagal trunks are divided at the oesophageal hiatus. This abolishes the pyloric relaxation reflex → must add a drainage procedure (pyloroplasty or gastrojejunostomy). In highly selective vagotomy, the nerve of Latarjet is preserved so pyloric function is maintained and no drainage is needed — but the operation is technically difficult [3].
Understanding PUD requires understanding the balance between aggressive and protective factors:
Aggressive factors ("Offence"):
- Hydrochloric acid (HCl) — secreted by parietal cells, stimulated by:
- Gastrin (from G cells in antrum) — endocrine pathway
- Histamine (from enterochromaffin-like [ECL] cells in corpus) — paracrine pathway via H₂ receptors
- Acetylcholine (from vagal nerve endings) — neural pathway via M₃ receptors
- Pepsin — secreted as inactive pepsinogen by chief cells; activated by acid (pH < 2); digests proteins including mucosal tissue
- Bile acids — refluxed bile can disrupt the mucosal barrier (relevant in post-gastrectomy states)
Defensive factors ("Defence"):
- Mucus-bicarbonate barrier — surface mucous cells secrete a thick gel layer of mucus + bicarbonate ions, creating a pH gradient from ~2 in the lumen to ~7 at the epithelial surface
- Prostaglandins (especially PGE₂ and PGI₂) — produced via COX-1 in mucosal epithelial cells; prostaglandins:
- Stimulate mucus and bicarbonate secretion
- Maintain mucosal blood flow (vasodilation)
- Promote epithelial cell turnover and repair
- Inhibit gastric acid secretion
- Mucosal blood flow — washes away back-diffused H⁺ ions and delivers oxygen/nutrients for repair
- Epithelial cell turnover — the gastric epithelium renews every 3–5 days; rapid restitution after injury
- Surface phospholipids — create a hydrophobic layer that repels acid
PUD occurs when aggressive factors overwhelm defensive factors.
The Ulcer Equation
PUD = ↑ Aggression (acid, pepsin, bile, H. pylori, NSAIDs) + ↓ Defence (mucus, bicarbonate, blood flow, prostaglandins, epithelial repair)
No acid, no ulcer — Karl Schwarz's dictum (1910). While this is an oversimplification (most DU patients have normal acid levels), acid is a necessary cofactor for virtually all peptic ulcers.
4. Etiology & Pathophysiology
H. pylori is the single most important aetiological factor worldwide.
Microbiology:
- Microaerophilic, Gram-negative, curved/spiral-shaped rod (coccobacillus)
- Possesses flagella → motility through the viscous mucus layer
- Produces mucolytic enzymes → facilitates penetration to the epithelial surface
- Has strong urease activity → this is vital for survival and colonisation:
- Bacterial urease hydrolyses gastric luminal urea → ammonia (NH₃) + CO₂
- Ammonia neutralises surrounding gastric acid, creating a protective alkaline cloud around the organism
- This is the basis for the rapid urease test (CLO test) and the urea breath test (UBT) used diagnostically [2]
Pathogenesis of DU vs GU — the two patterns of H. pylori gastritis:
| Feature | Antral-predominant gastritis → Duodenal ulcer | Pangastritis (antrum + body) → Gastric ulcer |
|---|---|---|
| Inflammation pattern | Confined to antrum; body (corpus) is spared | Involves both antrum and body |
| Effect on acid | ↑ Acid secretion (body parietal cells are intact and uninhibited; antral inflammation ↑ gastrin release and ↓ somatostatin) | ↓ Acid secretion (parietal cell destruction/atrophy in the body → hypochlorhydria) |
| Mechanism of ulcer | Excess acid overwhelms duodenal defences → DU | Mucosal defence is impaired by widespread inflammation despite lower acid → GU |
| Gastric cancer risk | Low (no atrophy, no metaplasia) | High (atrophy → intestinal metaplasia → dysplasia → carcinoma sequence) [2][4] |
Why does antral-predominant gastritis increase acid?
- H. pylori infection of antral G cells → ↑ gastrin release
- H. pylori infection of antral D cells → ↓ somatostatin (the "brake" on acid secretion is lost)
- Net effect: hypergastrinaemia → parietal cell hyperplasia in the (unaffected) body → acid hypersecretion
Why does pangastritis decrease acid?
- Inflammation extends to the body → direct damage to parietal cells → atrophic gastritis → loss of acid-secreting capacity
- Compensatory ↑ gastrin occurs but there are fewer functioning parietal cells to respond
Virulence factors of H. pylori:
- CagA (cytotoxin-associated gene A) — injected into host cells via a type IV secretion system; disrupts cell signalling, promotes inflammation, and is strongly associated with ulcers and gastric cancer
- VacA (vacuolating cytotoxin A) — induces vacuolation and apoptosis in epithelial cells
- Urease — as above, neutralises acid
- Flagella and adhesins (e.g., BabA, SabA) — enable colonisation
High Yield: H. pylori is found in ~90–95% of DU and ~60–80% of GU [2]. Eradication of H. pylori dramatically reduces ulcer recurrence from ~80% to < 5% per year.
NSAIDs are the second most common cause of PUD.
Pathogenesis:
-
Systemic prostaglandin inhibition (the main mechanism):
- Gastric and duodenal mucosa rely on COX-1 (constitutive) for the synthesis of prostaglandins (especially PGE₂ and PGI₂)
- Prostaglandins protect the mucosa by: mucin production, bicarbonate secretion, maintaining mucosal blood flow, inhibiting acid secretion, and promoting epithelial cell turnover
- Non-selective NSAIDs inhibit both COX-1 and COX-2 → impaired prostaglandin production → disruption of mucosal barrier → increased mucosal permeability to H⁺ ions → intramural acidosis → cell death → ulceration [2]
- This is a systemic effect — even parenteral or rectal NSAIDs cause gastric ulcers (not just direct topical injury)
-
Topical injury (minor contribution):
- Many NSAIDs are weak acids that are non-ionised in the acidic gastric lumen → they diffuse into mucosal cells where the neutral intracellular pH causes them to ionise and become trapped ("ion trapping") → direct cellular toxicity
-
Impaired platelet function:
- COX-1 inhibition in platelets → ↓ thromboxane A₂ → impaired platelet aggregation → if an ulcer forms, bleeding is more severe and harder to stop
Why selective COX-2 inhibitors (e.g., celecoxib, etoricoxib) are safer:
- COX-2 is mainly induced in inflammatory cells and is not the dominant isoform in gastric mucosa
- Selective COX-2 inhibition spares COX-1-dependent prostaglandin production in the stomach
- However, COX-2 inhibitors still carry some GI risk (COX-2 contributes to mucosal healing) and importantly increase cardiovascular risk (↓ PGI₂ in endothelium while TXA₂ production is preserved)
Risk factors for NSAID-related peptic ulcers [2]:
- Advanced age ( > 75 years)
- Prior history of clinical ulcer disease or ulcer complications
- High dose / long duration / relatively toxic NSAIDs
- Concurrent use of corticosteroids, anticoagulants, or antiplatelet agents
- Concomitant H. pylori infection (synergistic risk)
- Concurrent use of SSRIs (impair platelet serotonin uptake → worse bleeding)
NSAIDs + H. pylori — Synergistic Risk
The combination of NSAIDs and H. pylori infection has a synergistic (not just additive) effect on ulcer risk. Current guidelines recommend testing and eradicating H. pylori before initiating long-term NSAID therapy, especially in high-risk patients.
"Stress ulcers" occur in critically ill patients (burns, trauma, major surgery, sepsis, ICU patients on mechanical ventilation). This is physiological stress, not psychological stress.
Pathogenesis:
- Splanchnic hypoperfusion → mucosal ischaemia → breakdown of mucosal defence
- ↓ Mucosal blood flow → ↓ bicarbonate delivery → ↓ wash-out of back-diffused H⁺
- Reperfusion injury with free radical generation worsens mucosal damage
Eponymous types:
- Curling ulcer — associated with severe burns (named after Thomas Blizard Curling). Mechanism: hypovolaemia → splanchnic vasoconstriction → mucosal ischaemia. Typically in the duodenum.
- Cushing ulcer — associated with CNS injury/neurosurgery (named after Harvey Cushing). Mechanism: ↑ vagal tone from raised ICP → ↑ gastric acid secretion. Unique in that these ulcers have high acid as a driver. Typically deep, single ulcers that perforate.
- Impairs mucosal blood flow (nicotine → vasoconstriction)
- Reduces bicarbonate secretion from the pancreas and duodenal mucosa
- Promotes duodenogastric reflux of bile
- May enhance H. pylori colonisation
- Delays ulcer healing and increases recurrence
- Direct topical mucosal irritant → disrupts the mucosal barrier
- Stimulates acid secretion (especially at low concentrations)
- Causes acute gastritis (mucosal haemorrhage) rather than true chronic PUD per se, but exacerbates ulcers
A rare but important cause of refractory/unusual ulcers.
- Caused by a gastrinoma (gastrin-secreting neuroendocrine tumour) — most commonly in the duodenum (60–80%) or pancreas (20–40%), in the "gastrinoma triangle"
- Pathophysiology: Autonomous hypersecretion of gastrin → massive acid hypersecretion → overwhelms mucosal defences → multiple, refractory ulcers
- ↑ Gastrin also causes gastric epithelial cell (ECL cell and parietal cell) hyperplasia
When to suspect ZES [2]:
- Recurrent ulcers despite adequate treatment
- Ulcers in unusual locations (D2, jejunum)
- Complicated PUD without H. pylori or NSAID use
- Multiple ulcers simultaneously
- Ulcers associated with diarrhoea (massive acid inactivates pancreatic lipase → steatorrhoea; acid damages small bowel mucosa)
- Association with MEN1 (multiple endocrine neoplasia type 1) — parathyroid adenoma + pituitary adenoma + pancreatic/duodenal neuroendocrine tumour
Diagnosis: Fasting serum gastrin level (markedly elevated, typically > 1000 pg/mL) in the presence of high gastric acid output (to distinguish from the hypergastrinaemia of achlorhydria/PPI use, where gastrin rises as a compensatory response to low acid) [2].
- Corticosteroids — alone they have minimal ulcerogenic potential, but in combination with NSAIDs they significantly increase risk
- Crack cocaine, methamphetamines — mesenteric vasoconstriction → mucosal ischaemia
- Radiation — to upper abdomen
- Crohn's disease — can cause duodenal or gastric ulcers
- CMV/HSV — in immunocompromised patients (e.g. HIV/AIDS, post-transplant)
- Gastric cancer — a gastric ulcer may be malignant (always biopsy GU; DU is almost never malignant)
- Idiopathic ulcers — rising proportion; possibly related to mucosal hypersensitivity, visceral hyperalgesia, or unidentified pathogens
5. Classification
| Site | Frequency | Notes |
|---|---|---|
| Duodenal ulcer (DU) | ~75% | Almost always in D1 (duodenal bulb); >95% benign |
| Gastric ulcer (GU) | ~20% | Lesser curvature and antrum most common; must biopsy to exclude malignancy |
| Other | ~5% | Oesophagus (Barrett's), Meckel's diverticulum (ectopic gastric mucosa), surgical anastomoses (marginal/stomal ulcers) |
This classification is based on anatomical location and acid-secretory status and is particularly useful for guiding surgical management of GU.
| Type | Location | Acid Secretion | Surgical Approach [3] |
|---|---|---|---|
| Type I (most common, ~58%) | Body of stomach / lesser curvature near the angular incisure (incisura angularis) | Normal or ↓ | Distal gastrectomy + Billroth I/II |
| Type II (~22%) | Body of stomach (GU) + simultaneous duodenal ulcer | ↑↑ | Truncal vagotomy + antrectomy + Billroth II |
| Type III (~20%) | Prepyloric (within 3 cm of pylorus) | ↑↑ | Truncal vagotomy + antrectomy + Billroth II |
| Type IV (rare) | High on the lesser curvature near the GOJ | ↓↓ | Subtotal gastrectomy extending to include the ulcer + Billroth I/II/Roux-en-Y |
| Type V | Any location — medication-induced (NSAIDs) | Variable (usually normal) | Treat medically; surgery rarely needed |
Why Does This Classification Matter?
Types II and III are acid-hypersecretory states (behave more like DU) → surgery must include an acid-reducing procedure (vagotomy). Types I and IV are not acid-hypersecretory → surgery focuses on resecting the ulcer (to exclude malignancy and remove the diseased mucosa) rather than reducing acid [3].
This endoscopic classification grades the stigmata of recent haemorrhage (SRH) and predicts rebleeding risk — critical for guiding endoscopic therapy. Briefly:
| Forrest Class | Description | Rebleeding Risk |
|---|---|---|
| Ia | Spurting haemorrhage | ~90% |
| Ib | Oozing haemorrhage | ~50% |
| IIa | Non-bleeding visible vessel (NBVV) | ~40–50% |
| IIb | Adherent clot | ~20–30% |
| IIc | Flat pigmented spot | ~5–10% |
| III | Clean base ulcer | < 5% |
6. Clinical Features
A. Epigastric Pain — the cardinal symptom [2]
- Character: Gnawing, burning, or aching pain localised to the epigastrium.
- Pathophysiological basis: Exposed submucosal nerve endings in the ulcer crater are stimulated by gastric acid and pepsin contact; also local inflammation with release of inflammatory mediators (prostaglandins, bradykinin, substance P) activating visceral nociceptors.
- The relationship of pain to meals differs classically between DU and GU:
| Feature | Duodenal Ulcer | Gastric Ulcer |
|---|---|---|
| Pain timing | 2–5 hours after meals ("hunger pain"); relieved by eating or antacids | Within 30 min of eating; exacerbated by food |
| Night pain | Common (acid secretion peaks at ~2 AM via vagal drive during sleep) | Less common |
| Mechanism | Food buffers acid → pain relief; when stomach empties, unbuffered acid washes into D1 → pain returns | Food stimulates acid secretion and gastric distension → irritates ulcer |
| Weight | May gain weight (eating relieves pain) | May lose weight (eating worsens pain → food avoidance) |
- Periodicity: PUD pain classically waxes and wanes over weeks to months, with symptom-free intervals. This reflects cycles of ulcer healing and relapse.
- Radiation to the back: Atypical but important — suggests posterior penetration (DU penetrating into the pancreas; GU penetrating into the pancreatic body). Persistent back pain in PUD is a red flag [2].
Meal-Pain Relationship — Why?
In DU: food enters the stomach → acid is buffered → pain relief. But 2–5 hours later, the stomach empties and a bolus of unbuffered acid enters D1 → pain returns. This is why DU causes "hunger pain" and night pain (the stomach is empty, acid is unbuffered, vagal drive peaks overnight).
In GU: food → gastrin release → ↑ acid secretion + gastric distension → direct irritation of the ulcer in the stomach wall → pain with meals.
B. Nausea and Vomiting
- More common in GU than DU.
- Pathophysiology: gastric mucosal inflammation → visceral afferent stimulation → nausea; if there is pyloric oedema or spasm from a peri-pyloric ulcer, gastric emptying is delayed → vomiting.
- Projectile, non-bilious vomiting of undigested food → suggests gastric outlet obstruction (GOO) due to chronic scarring/fibrosis at the pylorus [3].
C. Early Satiety and Postprandial Fullness
- Due to gastric inflammation, impaired accommodation (fundus fails to relax normally), or pyloric oedema causing delayed emptying.
D. Belching and Bloating
- Aerophagia from repeated swallowing (a reflex response to nausea/pain); also gaseous distension from delayed emptying.
E. Anorexia and Weight Loss
- More common with GU (food avoidance due to pain).
- Must always exclude gastric malignancy in a patient with GU + weight loss.
F. Fatty Food Intolerance
- Fat delays gastric emptying and stimulates cholecystokinin (CCK) → may exacerbate symptoms.
G. Heartburn (Not a Primary Symptom of PUD)
- Heartburn (retrosternal burning) is more suggestive of GERD but may coexist — the two conditions share risk factors [2].
H. Symptoms of Complications (presenting features in some patients):
- Haematemesis / melaena → haemorrhage (the most common complication presenting acutely)
- Sudden severe epigastric pain ("thunderclap") radiating to the whole abdomen → perforation
- Persistent vomiting of old food, succussion splash → gastric outlet obstruction
High Yield: Up to 70% of NSAID-related ulcers are asymptomatic — the first presentation may be a life-threatening haemorrhage or perforation. This is because NSAIDs also have analgesic properties that mask ulcer pain [2].
Silent Ulcers
Do NOT assume a patient without epigastric pain does not have PUD. Elderly patients and those on NSAIDs frequently present with a complication (bleeding, perforation) as the first manifestation — the so-called "silent ulcer." Always consider PUD in elderly patients with unexplained anaemia or melaena even without pain.
A. General Examination — Often Normal in Uncomplicated PUD
In uncomplicated PUD, physical examination may be entirely unremarkable. This is important to recognise: the diagnosis of uncomplicated PUD is clinical (history) + endoscopic, not based on physical signs.
B. Epigastric Tenderness
- Mild, localised epigastric tenderness on palpation — the most common (but non-specific) sign.
- Pathophysiology: visceral inflammation in the upper GI tract → referred somatic pain/tenderness in the T6–T8 dermatome distribution (epigastrium).
- Absence of tenderness does NOT exclude PUD; its presence does not confirm it.
C. Signs of Complications:
| Complication | Signs | Pathophysiological Basis |
|---|---|---|
| Haemorrhage | Pallor, tachycardia, hypotension, postural drop, melaena on PR exam, haematemesis or coffee-ground aspirate on NG tube | Ulcer erodes into submucosal or serosal artery (e.g. GDA in posterior DU) → blood loss → hypovolaemic shock |
| Perforation | Board-like rigidity, guarding, rebound tenderness, absent bowel sounds, loss of liver dullness on percussion (Jobert sign — pneumoperitoneum), fever, tachycardia, shock | Free perforation → gastric/duodenal contents spill into peritoneal cavity → chemical then bacterial peritonitis → peritoneal irritation → reflex muscle spasm (rigidity) |
| Gastric outlet obstruction (GOO) | Succussion splash (audible splash on shaking the abdomen > 4 hours after last meal), visible gastric peristalsis, distended upper abdomen, dehydration, hypochloraemic hypokalaemic metabolic alkalosis | Chronic scarring at pylorus/D1 → mechanical obstruction → gastric dilation → repeated vomiting of HCl-rich gastric juice → loss of H⁺ and Cl⁻ → the kidneys compensate by retaining H⁺ and excreting K⁺ and HCO₃⁻, but the loss of Cl⁻ limits the kidney's ability to excrete HCO₃⁻ → alkalosis perpetuates [3] |
| Penetration | Persistent back pain (no longer relieved by food/antacids), signs of pancreatitis (if penetrating into pancreas) | Ulcer erodes posteriorly through the full thickness of the wall into an adjacent organ (pancreas, lesser omentum, liver) without free perforation (because the adjacent organ "seals" the hole) |
GOO Metabolic Derangement — Step by Step
- Vomiting loses HCl (H⁺ + Cl⁻) → metabolic alkalosis + hypochloraemia.
- Volume depletion → kidney activates RAAS → aldosterone → Na⁺ reabsorption in exchange for K⁺ and H⁺ secretion → hypokalaemia and paradoxical aciduria ("paradoxical" because the body is alkalotic but the kidneys are excreting acid to retain Na⁺).
- Loss of Cl⁻ means the kidney cannot excrete HCO₃⁻ (because Cl⁻ is needed in exchange for HCO₃⁻ reabsorption in the proximal tubule) → alkalosis is maintained = "chloride-responsive alkalosis."
- Treatment: IV normal saline (0.9% NaCl) repletes volume and chloride → kidneys can now excrete excess HCO₃⁻ → alkalosis corrects. Add KCl for hypokalaemia.
D. Signs Suggesting Alternative Diagnoses (Important Negatives):
- Jaundice → think biliary disease, pancreatic head malignancy, hepatocellular disease
- Lymphadenopathy (Virchow's node — left supraclavicular) → gastric malignancy
- Palpable mass → gastric cancer, not uncomplicated PUD
- Hepatomegaly / ascites → consider liver disease with portal hypertensive gastropathy or variceal bleeding mimicking PUD
These features warrant urgent upper endoscopy (OGD) to exclude malignancy:
- Age ≥ 55 with new-onset dyspepsia
- Family history of upper GI cancer
- Unintended weight loss
- GI bleeding (haematemesis, melaena, iron-deficiency anaemia)
- Progressive dysphagia or odynophagia
- Persistent vomiting
- Jaundice
- Palpable mass or lymphadenopathy
| Feature | Duodenal Ulcer | Gastric Ulcer |
|---|---|---|
| Location | D1 (bulb) | Lesser curvature, antrum |
| H. pylori association | ~90–95% | ~60–80% |
| Acid secretion | ↑ or normal | Normal or ↓ |
| Gastritis pattern | Antral-predominant | Pangastritis |
| Pain relation to food | Relieved by food; "hunger pain," night pain | Worsened by food |
| Weight | May gain | May lose |
| Malignancy risk | Essentially zero | ~5% of GU may be malignant → always biopsy |
| Cancer sequence | Not associated with gastric cancer | Atrophy → intestinal metaplasia → dysplasia → carcinoma |
| Perforation | Anterior wall → free peritoneum | Can also perforate (lesser sac) |
| Haemorrhage | Posterior wall → GDA | Lesser curvature → left gastric artery |
Always Biopsy Gastric Ulcers
Duodenal ulcers are almost never malignant and do not routinely need biopsy. Gastric ulcers MUST be biopsied (multiple biopsies from the ulcer rim) to exclude gastric adenocarcinoma or lymphoma. A non-healing GU after > 3 months of treatment requires repeat OGD + biopsy and strong consideration for surgical excision [3].
High Yield Summary
- PUD = mucosal defect through the muscularis mucosae — "no acid, no ulcer" but the balance of aggression vs defence is the true paradigm.
- DU (~75%) > GU (~20%); DU almost always in D1; GU on lesser curvature/antrum.
- Two dominant causes: H. pylori (90–95% DU, 60–80% GU) and NSAIDs — these are synergistic.
- NSAIDs cause ulcers systemically by inhibiting COX-1 → ↓ prostaglandins → ↓ mucus, bicarbonate, blood flow.
- Modified Johnson classification (Types I–V) classifies GU by location and acid status → guides surgical approach.
- "Posterior bleeds, Anterior perforates" — posterior DU erodes into GDA; anterior DU perforates freely.
- DU pain: hunger pain, relieved by food, nocturnal. GU pain: worse with food.
- Up to 70% of NSAID ulcers are silent — first presentation may be haemorrhage or perforation.
- Always biopsy GU to exclude malignancy; DU biopsy is not routine.
- GOO → non-bilious projectile vomiting, succussion splash, hypochloraemic hypokalaemic metabolic alkalosis.
- ZES: suspect if recurrent/refractory ulcers, unusual location (D2/jejunum), no H. pylori/NSAIDs.
- PUD is the most common cause of UGIB [1].
Active Recall - PUD Definition, Epidemiology, Anatomy, Etiology, Classification & Clinical Features
[1] Lecture slides: GC 198. Profuse vomiting of fresh blood and in shock severe upper GI bleeding.pdf (p7 — Causes of upper GI bleeding) [2] Senior notes: felixlai.md (Peptic ulcer disease sections, pp. 386–389; Dyspepsia section, pp. 327–328; Gastric cancer risk factors, p. 402) [3] Senior notes: maxim.md (Benign diseases of stomach — PUD, PUD complications, UGIB sections, pp. 52–53, 127, 130) [4] Senior notes: felixlai.md (Gastric cancer, p. 402 — relationship of H. pylori gastritis patterns to gastric cancer risk)
Differential Diagnosis of Peptic Ulcer Disease
When a patient presents with epigastric pain — the cardinal symptom of PUD — your job is not to jump straight to "peptic ulcer." The epigastrium is a crossroads: the stomach, duodenum, pancreas, biliary tree, oesophagus, and even the heart all refer pain here. A systematic differential diagnosis ensures you don't miss a life-threatening mimic and guides your investigation strategy.
The approach below is organised by the presenting complaint a PUD patient might have, because PUD presents in three main clinical scenarios:
- Dyspepsia / epigastric pain (most common — the "uncomplicated" presentation)
- Upper GI bleeding (haematemesis / melaena — the most common complication)
- Acute abdomen (perforation, peritonitis)
1. Differential Diagnosis of Dyspepsia / Epigastric Pain
This is the presentation where the patient walks into clinic or ED with epigastric discomfort, burning, postprandial fullness, or early satiety — and you are considering PUD as one of many possibilities.
| Diagnosis | Key Distinguishing Features | Why It Mimics PUD |
|---|---|---|
| Functional dyspepsia (~60% of all dyspepsia) [2] | Symptoms ≥ 3 months, onset ≥ 6 months ago; NO structural disease on OGD; diagnosis of exclusion | Identical symptom profile (epigastric pain/burning, early satiety, postprandial fullness). The difference is that OGD is normal — there is no mucosal break |
| Gastritis / Duodenitis (erosive) [1][3] | Drug-induced (aspirin/NSAIDs), alcohol-induced, stress-induced; bleeding is typically self-limited; diagnosis by OGD showing mucosal erythema/erosions without a true ulcer | Erosions cause similar burning pain and can bleed, but they do not breach the muscularis mucosae — i.e., they are superficial |
| Gastric malignancy (adenocarcinoma, lymphoma, GIST) [3][5] | Constitutional symptoms (weight loss, anorexia, cachexia); dysphagia, early satiety, persistent epigastric pain; palpable mass; Virchow's node; anaemia | A malignant gastric ulcer can look identical to a benign GU at endoscopy — this is why all GU must be biopsied |
| Dieulafoy's lesion [3] | Idiopathic dilated aberrant submucosal vessel eroding normal overlying mucosa; MC in gastric fundus; presents with acute UGIB rather than chronic pain | Presents as UGIB, can be missed on OGD if not actively bleeding |
| Gastric outlet obstruction [3] | Non-bilious projectile vomiting of undigested food, succussion splash; malignant until proven otherwise (80% malignant — CA stomach MC, CA head of pancreas; 20% benign — PUD-related pyloric stenosis is 2nd MC) | GOO can be a complication of PUD itself, but also caused by periampullary/pancreatic malignancy |
| Gastric volvulus [3] | Borchardt's triad: severe epigastric pain, retching without vomiting, inability to pass NG tube; usually secondary to rolling hiatal hernia | Acute upper abdominal pain mimicking perforated PUD |
| Diagnosis | Key Distinguishing Features | Why It Mimics PUD |
|---|---|---|
| Gastro-oesophageal reflux disease (GERD) [2] | Retrosternal burning (heartburn), regurgitation, worse lying flat/bending; relieved by antacids/PPIs; water brash | Heartburn can be perceived as "epigastric pain"; both respond to PPIs, causing diagnostic confusion |
| Oesophagitis / oesophageal ulcers (erosive, infective, pill) [1] | Odynophagia; Hx of GERD, immunosuppression (HSV, Candida), or causative medications (bisphosphonates, tetracycline, KCl) | Retrosternal/epigastric pain; can cause UGIB |
| Oesophageal malignancy | Progressive dysphagia (solids → liquids), weight loss, anaemia | Late-stage CA oesophagus can cause epigastric pain and UGIB |
| Diagnosis | Key Distinguishing Features | Why It Mimics PUD |
|---|---|---|
| Biliary colic [6] | RUQ/epigastric pain after fatty meals, lasting 30 min–6 hours then resolving completely; no fever, no peritoneal signs | Epigastric component can be mistaken for PUD; but biliary colic is colicky (waxes/wanes), resolves spontaneously, and is precipitated specifically by fatty meals |
| Acute cholecystitis [6] | RUQ pain > 6 hours, fever, Murphy's sign, ↑ WCC, ↑ CRP | Pain more RUQ than epigastric; peritoneal signs present; does NOT resolve spontaneously (unlike biliary colic and unlike DU pain which is relieved by food) |
| Acute pancreatitis [7] | Epigastric pain radiating to the back, relieved by sitting up/leaning forward; nausea/vomiting; ↑ amylase/lipase ( > 3× ULN); Hx of gallstones or alcohol | The back radiation and posture relief are key distinguishing features. PUD can also radiate to the back (posterior penetration into the pancreas), but pancreatitis pain is more constant and severe |
| Chronic pancreatitis [2] | Chronic epigastric pain radiating to the back; steatorrhoea; weight loss; pancreatic calcifications on imaging | Chronic pain mimicking chronic PUD; Hx of alcohol use or recurrent acute pancreatitis |
| Pancreatic / periampullary malignancy | Painless obstructive jaundice; weight loss; Courvoisier sign; new-onset diabetes | Late-stage disease causes epigastric pain |
| Diagnosis | Key Distinguishing Features | Why It Mimics PUD |
|---|---|---|
| Acute myocardial infarction (especially inferior MI) [5][7] | Chest tightness/pressure, diaphoresis, dyspnoea; ECG changes; troponin elevation; risk factors (DM, HTN, smoking, hyperlipidaemia) | Inferior MI (RCA territory) can present as isolated epigastric pain with nausea/vomiting — the diaphragmatic surface of the heart refers pain to the epigastrium via the phrenic nerve and shared T5–T9 dermatomes. Always do an ECG in any patient > 50 with acute epigastric pain |
| Pericarditis | Pleuritic chest pain worse lying flat, relieved sitting forward; pericardial rub; diffuse ST elevation | Epigastric component may dominate |
Don't Kill Your Patient
A middle-aged patient presenting to ED with acute epigastric pain, nausea, and diaphoresis could have an inferior MI, not just a peptic ulcer. Always get an ECG before attributing epigastric pain to a GI cause, especially in patients with cardiovascular risk factors. This is a classic exam trap and a real-life killer.
| Diagnosis | Key Distinguishing Features |
|---|---|
| Medications [2] | NSAIDs, glucocorticoids, ampicillin, erythromycin, iron supplements, K⁺ supplements, bisphosphonates — can cause dyspepsia without frank ulceration |
| Metabolic [2] | Hypercalcaemia (stimulates gastrin → ↑ acid; also causes constipation, "stones, bones, groans, moans"); hyperkalaemia |
| Diabetic ketoacidosis (DKA) [5] | Abdominal pain, nausea/vomiting, Kussmaul breathing, fruity breath, hyperglycaemia, ketones |
| Mesenteric ischaemia [5] | "Pain out of proportion to examination"; post-prandial pain ("intestinal angina"); AF, vascular risk factors |
| Irritable bowel syndrome | Chronic abdominal pain relieved by defecation; altered bowel habit; no alarm features |
When PUD bleeds, it enters the differential of all causes of upper GI bleeding. The lecture slides list these in descending order of frequency [1]:
Causes of upper GI bleeding in descending order of frequency [1]:
- Duodenal or gastric ulcer (most common)
- Gastritis
- Esophageal or gastric varices
- Mallory-Weiss syndrome
- Benign or malignant gastric tumour
Expanding on each, organised by anatomical site [1][3][4]:
Oesophageal causes:
- Oesophagitis (erosive — GERD-related, infective, pill-induced)
- Oesophageal varices — complication of portal hypertension / liver cirrhosis; presents with massive haematemesis; look for stigmata of chronic liver disease (spider naevi, caput medusae, ascites, jaundice) [4]
- Mallory-Weiss syndrome — longitudinal mucosal laceration at the GOJ following forceful retching/vomiting; the history of preceding forceful vomiting followed by haematemesis is the key clue (contrast with Boerhaave syndrome which is a full-thickness perforation) [1][3]
- Oesophageal malignancy
Gastric causes:
- PUD — most common cause [1]
- Gastritis (drug-induced, alcohol-induced, stress-induced) — inflammation without a true ulcer; bleeding typically self-limited [1]
- Portal hypertensive gastropathy — friable mucosa with mosaic "snakeskin" pattern on OGD; bleeding from ectatic mucosal vessels rupturing; associated with portal hypertension [1][4]
- Gastric varices — often fundal; more difficult to manage endoscopically than oesophageal varices
- Dieulafoy's lesion — aberrant dilated submucosal artery; MC in gastric fundus; can cause massive bleeding from a normal-appearing mucosal surface [3]
- Gastric malignancy (adenocarcinoma, lymphoma, GIST) [1]
Duodenal causes:
- Duodenal ulcer (part of PUD)
- Duodenitis
- Duodenal malignancy
- Haemobilia (bleeding from the biliary tree into the duodenum — associated with cholangiocarcinoma, liver biopsy, hepatic artery aneurysm) [3]
Other:
- Aorto-enteric fistula — must consider in any patient with a history of aortic graft repair who presents with UGIB; classically a "herald bleed" (small initial bleed) followed by catastrophic exsanguination; CT aortogram is diagnostic [3][4]
- Angiodysplasia / AV malformation — dilated, tortuous submucosal vessels; can occur anywhere but often in stomach and duodenum in the upper GIT; diagnosed endoscopically [1]
- Haemosuccus pancreaticus — bleeding from the pancreatic duct, usually in patients with chronic pancreatitis, pseudocysts, or pancreatic tumours [1]
Mnemonic — UGIB Differential by Site
"VEGaS MaD" — Varices, Erosive oesophagitis/gastritis, Gastric ulcer, Stomach tumour, Mallory-Weiss, angioDysplasia. Plus always think PUD (most common) and aorto-enteric fistula (most dangerous to miss).
Perforated peptic ulcer (PPU) presents as sudden-onset severe generalised abdominal pain with peritonism. The differential overlaps with the life-threatening causes of acute abdomen [5]:
- Perforated viscus (PPU is most common; also perforated appendix, diverticular perforation, colonic perforation)
- Ruptured AAA — pulsatile abdominal mass, back pain, hypotension; in an elderly male with known AAA
- Acute mesenteric ischaemia — pain out of proportion to examination; AF; metabolic acidosis
- Acute intestinal obstruction
- Severe acute pancreatitis
- Ruptured HCC — in patient with known chronic liver disease
- Medical causes: DKA, acute MI, Addisonian crisis [5]
- Obstetric causes: ruptured ectopic pregnancy, placental abruption [5]
A specific clinical pearl for appendicitis: Valentino's sign — gastric/duodenal content from a PPU tracks down the right paracolic gutter to the RIF, mimicking acute appendicitis. The clue is that pain started in the epigastrium before migrating to the RIF (in appendicitis, pain starts peri-umbilical then migrates to the RIF, but the initial pain is visceral and vague, not the "thunderclap" epigastric pain of PPU) [5].
The history alone can narrow down the differential substantially. Here are the key discriminating questions:
Relationship to food:
- DU: pain relieved by food, recurs 2–5 hours later ("hunger pain") [2][5]
- GU: pain worsened by food [2][5]
- Biliary colic: pain precipitated by fatty meals specifically [5]
- Pancreatitis: pain worsened by food, relieved by leaning forward [5]
- Functional dyspepsia: variable relationship, no consistent pattern
Character and radiation:
- PUD: burning/gnawing epigastric pain; back radiation if penetrating posteriorly [2]
- Pancreatitis: band-like epigastric pain radiating to back [7]
- Biliary colic: RUQ/epigastric pain radiating to right shoulder/scapula
- MI: crushing/pressure chest pain radiating to jaw/left arm; diaphoresis
Red flags mandating urgent OGD [2]:
- Age ≥ 55 with new dyspepsia
- Weight loss, dysphagia, odynophagia
- GI bleeding or iron-deficiency anaemia
- Persistent vomiting
- Palpable mass or lymphadenopathy
- Family history of upper GI cancer
- Jaundice
The 'Test and Treat' Strategy
In patients < 55 without alarm features, current guidelines recommend a "test and treat" approach: non-invasive H. pylori testing (urea breath test or stool antigen), and if positive, eradication therapy. If negative, an empirical PPI trial. OGD is reserved for those who fail initial management or have alarm features. This avoids unnecessary endoscopy in the majority of young dyspeptic patients [2].
| Presentation | Top Differentials to Consider | "Must Not Miss" |
|---|---|---|
| Epigastric pain / dyspepsia | Functional dyspepsia, GERD, gastritis, biliary colic, pancreatitis, gastric malignancy | Inferior MI, gastric cancer |
| UGIB | PUD (MC), gastritis, oesophageal/gastric varices, Mallory-Weiss, gastric tumour | Aorto-enteric fistula (Hx of aortic graft), variceal bleed (liver disease) |
| Acute abdomen (perforation) | PPU, ruptured AAA, mesenteric ischaemia, acute pancreatitis, ruptured HCC | Ruptured AAA, mesenteric ischaemia, MI |
| GOO | Gastric cancer (MC, 80%), PUD-related pyloric stenosis (2nd MC benign cause), CA head of pancreas | Malignant GOO — malignant until proven otherwise [3] |
High Yield Summary
- PUD is the most common cause of UGIB — but always consider varices (liver disease), Mallory-Weiss (Hx of forceful vomiting), malignancy (constitutional symptoms), and aorto-enteric fistula (Hx of aortic graft).
- Functional dyspepsia accounts for ~60% of dyspepsia — diagnosis of exclusion after OGD is normal.
- Always do an ECG in acute epigastric pain to exclude inferior MI — the single most dangerous mimic.
- Gastric ulcers must always be biopsied to exclude malignancy — a malignant gastric ulcer looks identical to a benign one.
- GOO is malignant until proven otherwise (80% malignant, 20% benign).
- Valentino's sign: PPU fluid tracking to RIF mimicking appendicitis.
- Aorto-enteric fistula: any patient with Hx of aortic graft + UGIB → CT aortogram urgently.
- "Test and treat" strategy for young patients without alarm features — non-invasive H. pylori testing before endoscopy.
- UGIB differentials by site: Oesophagus (varices, oesophagitis, Mallory-Weiss, CA), Stomach (PUD, gastritis, Dieulafoy, portal hypertensive gastropathy, varices, CA), Duodenum (DU, duodenitis, haemobilia, CA).
Active Recall - Differential Diagnosis of PUD
References
[1] Lecture slides: GC 198. Profuse vomiting of fresh blood and in shock severe upper GI bleeding.pdf (p7 — Causes of upper GI bleeding in descending order of frequency) [2] Senior notes: felixlai.md (Dyspepsia sections pp. 327–330; PUD sections pp. 386–390; UGIB differential diagnosis pp. 334–337) [3] Senior notes: maxim.md (UGIB differential diagnosis p. 52; Benign diseases of stomach — PUD, GOO, Dieulafoy, gastric volvulus pp. 127–132) [4] Senior notes: felixlai.md (Portal hypertensive gastropathy and variceal haemorrhage pp. 450–451) [5] Senior notes: maxim.md (Acute abdomen differential diagnosis pp. 43–44; Appendicitis differential — Valentino's sign p. 180) [6] Senior notes: felixlai.md (Biliary colic vs acute cholecystitis differential pp. 555) [7] Senior notes: felixlai.md (Acute pancreatitis — clinical manifestation and differential diagnosis pp. 579–580)
Diagnostic Criteria, Diagnostic Algorithm & Investigation Modalities for Peptic Ulcer Disease
Unlike conditions such as rheumatoid arthritis or heart failure, PUD does not have a set of formal "diagnostic criteria" with point scores. Instead, the diagnosis rests on a straightforward principle:
The gold standard for diagnosing PUD is direct visualisation of the ulcer on oesophago-gastro-duodenoscopy (OGD) with biopsy [2][3].
That said, the clinical approach follows a structured algorithm driven by two key questions:
- Does this patient need an OGD now, or can we test-and-treat? (determined by the presence or absence of alarm features)
- Once an ulcer is found, what is the aetiology? (determined by H. pylori testing, drug history, and biopsy findings)
Additionally, when PUD presents as a complication — UGIB, perforation, or GOO — the diagnostic pathway shifts to an emergency investigation algorithm (covered below).
The following algorithm integrates the approach to dyspepsia/suspected PUD and the emergency PUD complication pathways into one unified flowchart.
The Two Gatekeeper Questions
Question 1 — Is this an emergency? If the patient has UGIB, peritonism, or GOO → emergency pathway (resuscitate first, investigate second).
Question 2 — Are there alarm features? If yes → straight to OGD. If no and age < 55 → "test and treat" with non-invasive H. pylori testing. This avoids unnecessary endoscopy in the majority of young dyspeptic patients [2].
These "red flags" indicate possible malignancy or serious pathology — bypass the "test and treat" approach and proceed directly to endoscopy:
- Age ≥ 55 with new-onset dyspepsia
- Family history of upper GI cancer
- Jaundice
- Unintended weight loss
- Dysphagia
- Odynophagia
- GI bleeding (haematemesis, melaena, haematochezia)
- Unexplained iron-deficiency anaemia
- Persistent vomiting
- Palpable mass or lymphadenopathy [2]
High Yield: The alarm features are commonly tested. Remember them by the mnemonic "VBAD FLaG" — Vomiting (persistent), Bleeding (GI), Anaemia (IDA), Dysphagia/odynophagia, Family history of upper GI cancer, Loss of weight, age ≥ 55, Gland (lymphadenopathy/mass).
4. Investigation Modalities — Detailed Breakdown
We now cover every investigation relevant to PUD, organised into bedside tests → blood tests → H. pylori testing → imaging → endoscopy → special tests, with the rationale and key findings for each.
| Test | What It Tells You | Key Findings in PUD |
|---|---|---|
| Vital signs | Haemodynamic status — detect shock from haemorrhage | Tachycardia, hypotension, postural drop (Class II–IV haemorrhagic shock) |
| PR examination | Detect melaena or haematochezia | Tarry black stool on glove = melaena → suggests UGIB. Fresh blood = consider massive UGIB or LGIB |
| Urinalysis [8] | Exclude urological causes of abdominal pain | Normal in PUD. Haematuria → ureteric colic; nitrites/leucocytes → UTI |
| Pregnancy test [8] | Exclude ectopic pregnancy in women of childbearing age presenting with abdominal pain | Must be done in all females of reproductive age with acute abdomen |
| NG tube aspirate (if UGIB suspected) | Confirm upper GI source; assess ongoing bleeding | Coffee-ground or bloody aspirate confirms UGIB. Clear aspirate does NOT exclude UGIB (pyloric spasm can prevent reflux of duodenal blood into the stomach) |
| Test | Rationale | Key Findings / Interpretation |
|---|---|---|
| CBC with differentials [2][8] | Baseline Hb; detect anaemia; WCC for infection/inflammation | Microcytic hypochromic anaemia → iron-deficiency anaemia from chronic occult blood loss [2]. Note: in acute haemorrhage, initial Hb may be normal because both plasma and red cells are lost simultaneously — Hb drops only after haemodilution (fluid shifts or IV fluid resuscitation) |
| Clotting profile (PT/INR, APTT) [8] | Coagulopathy assessment; guide transfusion | Prolonged in liver disease, warfarin use, DIC. Affects management of bleeding ulcer |
| Type and screen / cross-match [8] | Prepare for blood transfusion | Always group & save in UGIB; cross-match 2–4 units if actively bleeding |
| LRFT (liver + renal function) [5][8] | Liver disease (portal hypertension → variceal DDx); renal function for contrast safety and drug dosing | Elevated urea:creatinine ratio ( > 100:1) → classic for UGIB. Why? Haemoglobin in the GI tract is digested → amino acids are absorbed → hepatic urea synthesis ↑. Also, hypovolaemia → ↓ renal perfusion → ↑ urea reabsorption. Creatinine is unaffected → ratio rises [5] |
| Amylase / lipase [8] | Exclude acute pancreatitis (key differential) | > 3× ULN suggests pancreatitis rather than PUD. However, a perforated posterior DU penetrating the pancreas can also mildly elevate amylase |
| CRP [8] | Inflammatory marker; raised in perforation, peritonitis | Non-specific but supports inflammatory/infective process |
| ABG + lactate [8] | Assess shock severity; detect metabolic acidosis from tissue hypoperfusion; detect GOO metabolic picture | Metabolic acidosis (raised lactate) → haemorrhagic shock or mesenteric ischaemia. Hypochloraemic hypokalaemic metabolic alkalosis → GOO from prolonged vomiting [3] |
| Glucose [8] | Exclude DKA (epigastric pain differential) | Hyperglycaemia + ketoacidosis = DKA |
| Calcium / phosphate [8] | Hypercalcaemia can cause dyspepsia and also stimulates gastrin → acid ↑; associated with MEN1 (ZES) | ↑ Ca²⁺ → think MEN1 (hyperparathyroidism + ZES + pituitary adenoma) |
| ECG + cardiac enzymes [8] | Exclude acute MI (inferior MI mimics PUD) | ST changes in leads II, III, aVF → inferior MI; troponin rise |
The Urea:Creatinine Ratio in UGIB
A urea:creatinine ratio > 100:1 (when both are in mmol/L) is a simple bedside clue that the bleeding source is upper GI. The mechanism is two-fold:
- Protein digestion: blood (haemoglobin = protein) is digested in the GI tract → amino acids absorbed → liver converts to urea → serum urea rises.
- Pre-renal AKI: hypovolaemia from blood loss → ↓ GFR → ↑ urea reabsorption in the proximal tubule (but creatinine is not reabsorbed). Both mechanisms selectively raise urea while creatinine stays relatively stable [5].
4.3 H. pylori Testing — The Cornerstone of Aetiological Diagnosis [2]
ALL patients diagnosed with peptic ulcer disease by endoscopy should undergo testing for H. pylori infection [2]. This is non-negotiable. The choice of test depends on whether an OGD is being performed.
| Test | Specimen | Mechanism | Sensitivity/Specificity | Key Points |
|---|---|---|---|---|
| Rapid urease test (CLO test) | 1 biopsy specimen | Biopsy placed in urea-containing medium with pH indicator. H. pylori urease cleaves urea → NH₃ → alkaline pH → colour change from yellow to pink [2] | Sens ~90–95%, Spec ~95–100% | Fast (result in 1–24 hours). First-line invasive test. False negatives if: recent PPI use (↓ bacterial load), recent antibiotics, active bleeding (blood dilutes the specimen) |
| Histological examination | 2 biopsy specimens | Microscopic identification of H. pylori organisms + assessment of mucosal pathology (gastritis, atrophy, intestinal metaplasia, dysplasia) [2] | Sens ~95%, Spec ~99% | Gold standard for detecting associated mucosal changes. Special stains (Giemsa, Warthin-Starry, immunohistochemistry) improve detection. Also identifies malignancy on GU biopsy |
| Bacterial culture + antibiotic sensitivity | Biopsy specimen | Culture in microaerophilic conditions | Sens ~70–80%, Spec 100% | Only indicated in treatment-refractory cases [2] — to guide antibiotic selection based on resistance patterns. Slow (takes days). Important in Hong Kong where clarithromycin resistance is rising (~20–30%) |
Biopsy site: Antrum is the preferred site because H. pylori density is highest there (the organism avoids acid-secreting parietal cells concentrated in the fundus/body) [2]. However, an additional biopsy from the gastric body should be taken to increase sensitivity, because H. pylori can migrate proximally following acid suppression therapy or partial eradication [2].
False Negatives in H. pylori Testing
PPIs, antibiotics, and bismuth must be stopped at least 2 weeks (PPIs) or 4 weeks (antibiotics/bismuth) before testing to avoid false negatives. PPIs raise intragastric pH → H. pylori shifts to a coccoid (dormant) form → reduced urease activity → negative CLO test/UBT. Active UGIB can also cause false negatives (blood dilutes the specimen and alkalinises the micro-environment).
| Test | Mechanism | Sensitivity/Specificity | Key Points |
|---|---|---|---|
| Urea breath test (UBT) | Patient ingests urea labelled with ¹³C (non-radioactive, more expensive) or ¹⁴C (radioactive, cheaper — avoid in pregnant women and young children). If H. pylori is present, its urease cleaves the labelled urea → labelled CO₂ is absorbed into the blood → exhaled and detected in breath samples [2] | Sens ~95–97%, Spec ~95–97% | Best non-invasive test for both initial diagnosis and confirmation of eradication. Must stop PPI ≥ 2 weeks before. ¹⁴C not preferred in pregnancy/children due to radiation |
| Stool antigen detection test (SAT) | Detects H. pylori antigens in stool using monoclonal antibodies | Sens ~92–95%, Spec ~92–95% | Good alternative to UBT; useful in children and when UBT is unavailable. Also valid for confirming eradication. Must stop PPI ≥ 2 weeks before |
| Serology (IgG antibodies) | Detects anti-H. pylori IgG antibodies in blood | Sens ~85–90%, Spec ~80–85% | Cannot distinguish active infection from past exposure — antibodies persist for months-years after eradication. Therefore NOT recommended for confirming eradication or for use in populations with declining H. pylori prevalence. Main use: epidemiological studies. Not affected by PPI/antibiotics |
| Urine antibody detection test [2] | Detects anti-H. pylori antibodies in urine | Lower accuracy than UBT/SAT | Convenient but less reliable; not widely used for clinical decisions |
Key principle for choosing the test:
- OGD being done? → CLO test + histology (always)
- No OGD needed (test-and-treat strategy)? → UBT (preferred) or SAT
- Confirming eradication post-treatment? → UBT (best) or SAT, performed ≥ 4 weeks after completing eradication therapy and ≥ 2 weeks after stopping PPI
- Serology → almost never the right answer in clinical practice (cannot confirm active infection or eradication)
4.4 Imaging Studies
Primary role: Detect pneumoperitoneum (free gas under the diaphragm) in suspected perforated peptic ulcer (PPU).
Mechanism: When an anterior ulcer perforates, gastric/duodenal gas escapes into the peritoneal cavity. Free gas rises to the highest point in an erect patient → collects under the diaphragm → visible as a crescent of radiolucency between the liver and the right hemidiaphragm (or under the left hemidiaphragm).
Key points:
- Sensitivity ~75–80% — so a negative erect CXR does NOT exclude perforation. If clinical suspicion remains, proceed to CT.
- As little as 1 mL of free gas can be detected on an erect CXR if the film quality is good.
- Also look for: left-sided pleural effusion (pancreatitis DDx), pneumomediastinum (Boerhaave syndrome DDx), cardiomediastinal abnormalities (cardiac DDx).
- Loss of liver dullness on percussion (Jobert sign) is the clinical correlate of pneumoperitoneum.
| Finding | Significance |
|---|---|
| Dilated gastric bubble | GOO — gastric distension from obstruction |
| Hourglass stomach (enlarged gastric bubble + dilated proximal duodenum + lack of distal small bowel gas) | Classic GOO appearance [3] |
| Air-fluid levels | If in small bowel → intestinal obstruction (DDx) |
| Pancreatic calcifications | Chronic pancreatitis (DDx for epigastric pain) |
| Calcified mass in RUQ | Bouveret syndrome (large gallstone impacted in duodenum causing GOO) [3] |
The workhorse investigation when CXR is negative but perforation is suspected, or when the clinical picture is unclear.
| Role | Key Findings |
|---|---|
| Confirm PPU when CXR is negative | Extraluminal free gas (even tiny amounts), discontinuity of bowel wall, localised fluid collection, mesenteric fat stranding |
| Diagnose complications | Penetration into pancreas (fat stranding around pancreas), abscess formation, GOO (transition point), aorto-enteric fistula |
| Exclude differential diagnoses | Pancreatitis (peripancreatic inflammation/necrosis), ruptured AAA (retroperitoneal haematoma), mesenteric ischaemia (bowel wall thickening, portal venous gas), malignancy |
| GOO assessment [3] | Better for identifying malignant causes (CA head of pancreas, periampullary tumour) |
When to Order CT vs Go Straight to Surgery
If the patient has florid peritoneal signs (board-like rigidity, rebound, absent bowel sounds) AND free gas on erect CXR → proceed directly to exploratory laparotomy without CT [5]. CT is for equivocal cases where the diagnosis is uncertain or to plan the surgical approach.
- NOT routinely done nowadays due to: (1) widespread OGD availability, (2) desire to limit radiation exposure, (3) inability to biopsy, (4) lower sensitivity than OGD [2].
- Historical significance: could demonstrate an ulcer crater as a "niche" (barium collecting in the ulcer) with surrounding mucosal folds radiating towards it (benign) vs irregular shouldering/mass effect (malignant).
- Current niche use: water-soluble contrast study (Gastrografin, not barium) may be used if perforation is suspected but CT is unavailable — barium is contraindicated in suspected perforation because barium peritonitis is lethal.
4.5 Oesophago-Gastro-Duodenoscopy (OGD) — The Gold Standard [2][3][9]
OGD is both diagnostic and therapeutic — this is the single most important investigation in PUD [2][9].
- Dyspepsia WITH alarm features → urgent OGD
- Age ≥ 55 with new-onset dyspepsia → OGD
- UGIB → urgent OGD within 24 hours once haemodynamically stabilised [1]
- Failure of test-and-treat strategy → OGD
- Follow-up of gastric ulcer → repeat OGD to confirm healing and exclude missed malignancy [2]
- GOO → OGD after NG decompression to biopsy and identify cause [3]
- Known or suspected perforation — gas insufflation during endoscopy can worsen a sealed-off perforation → free perforation → peritonitis [5]
- Recent myocardial infarction [9]
- Haemodynamic instability (relative) — stabilise first, then scope
AVOID Endoscopy in Acute Abdomen with Suspected Perforation
A sealed-off perforation may be converted to a free perforation by gas insufflation during endoscopy [5]. If peritonism and pneumoperitoneum are present, go straight to surgery — not the endoscopy suite.
A. Ulcer Morphology — Benign vs Malignant [2]
This is critically important because it guides whether a gastric ulcer needs biopsy (spoiler: it always does).
| Feature | Benign Ulcer | Malignant Ulcer |
|---|---|---|
| Edges | Smooth, regular, rounded [2] | Irregular, raised, thickened, or rolled [2] |
| Base | Flat, smooth, often filled with white/yellow fibrinous exudate [2] | Necrotic, irregular, may contain mass |
| Surrounding folds | Radiate symmetrically towards the crater (like spokes of a wheel) | Nodular, clubbed, fused, or amputated — folds do not radiate smoothly [2] |
| Mass effect | Absent | Ulcerated mass protruding into lumen [2] |
High Yield: Even if an ulcer "looks" benign endoscopically, you must biopsy all gastric ulcers — visual assessment alone misses ~5% of gastric cancers presenting as "benign-looking" ulcers. Take multiple biopsies from the ulcer rim (at least 4–6 from different quadrants) [2][3].
B. Forrest Classification — Stigmata of Recent Haemorrhage (SRH) [2]
When a bleeding ulcer is found, the endoscopic appearance predicts rebleeding risk and guides therapy.
| Forrest Class | Stigmata of Recent Haemorrhage | Prevalence | Rebleeding Risk | Action |
|---|---|---|---|---|
| Ia | Spurting haemorrhage | ~10% | 55–100% | Endoscopic therapy required |
| Ib | Oozing haemorrhage | ~10% | ~50% | Endoscopic therapy required |
| IIa | Non-bleeding visible vessel (NBVV) | ~25% | 40–50% | Endoscopic therapy required |
| IIb | Adherent clot | ~10% | 20–30% | Endoscopic therapy required (after vigorous flushing to reveal underlying vessel*) [3] |
| IIc | Flat pigmented spot | ~10% | ~10% | Low risk — PPI alone |
| III | Clean base | ~35% | ~5% | Low risk — start feeding, early discharge [1] |
* Need to remove adherent clot by vigorous flushing to reveal underlying vessels [3]
Key principle:
- High risk (Forrest I and IIa, IIb): Endoscopic therapy required + post-OGD IV PPI infusion [1][3]
- Low risk (Forrest IIc and III): Acid suppression alone is adequate — clean base: start feeding, early discharge [1]
C. Endoscopic Therapeutic Modalities for Bleeding Ulcers [1][3]
Dual therapy (combination of two modalities) is the standard for bleeding ulcers [3]:
| Modality | Mechanism | Details |
|---|---|---|
| Injection therapy: Adrenaline 1:10,000 [1][3] | Tamponade (volume effect compresses the vessel) + vasoconstriction + platelet aggregation | Injected in 4 quadrants around the bleeding point. Adrenaline alone is insufficient — must combine with a second modality |
| Thermal therapy: Heater probe [1][3] | Coaptive effect — simultaneous pressure (compresses the vessel) + heat (coagulates the vessel wall, sealing it) | Most effective thermal method. Apply firm pressure then activate heat |
| Mechanical therapy: Metal clip [1][3] | Physically grasps and closes the bleeding vessel | More prolonged action than thermal methods; useful for discrete vessels |
| Mechanical therapy: Haemospray [1][3] | Mechanical barrier + absorbent (absorbs water from blood → concentrates clotting factors) — "雲南白藥 principle" | Sprayed onto the bleeding surface; useful for large-area oozing where clips/thermal are impractical |
Why Dual Therapy?
Adrenaline injection alone reduces rebleeding by ~15–20%, but adding a second modality (thermal or mechanical) reduces it by ~60–70%. The adrenaline provides temporary haemostasis by tamponade and vasoconstriction, while the heater probe or clip provides definitive vessel sealing. This is why dual therapy (adrenaline + heater probe OR clip) is the standard [1][3].
D. Limitations of Endoscopic Haemostasis [3]
Endoscopy may fail when:
- Massive bleeding obscuring the view — cannot visualise the bleeding point
- Large bleeding artery ( > 3.2 mm diameter) — too large for clip or thermal coagulation
- Large ulcer ( > 2 cm) — difficult to access the bleeding vessel [3]
In these cases → surgical or interventional radiology (TAE) [3].
| Ulcer Type | Follow-Up OGD Needed? | Rationale |
|---|---|---|
| Gastric ulcer | Yes — always, until complete healing is confirmed | Risk of missed gastric cancer due to sampling error at initial biopsy. Healing of the ulcer reassures that the lesion is likely benign [2]. Repeat at 8–12 weeks |
| Uncomplicated duodenal ulcer | Not necessary if asymptomatic | Majority of DU are benign; malignancy risk is essentially zero [2] |
| Complicated duodenal ulcer | Yes — until complete healing confirmed | Ensure ulcer has healed to prevent recurrent complications [2] |
| Non-healing GU after 12 weeks | Mandatory — consider surgery even if biopsy is benign | Failure to heal despite adequate treatment raises strong suspicion for malignancy or ZES [2][3] |
High Yield: A non-healing gastric ulcer after 3 months (12 weeks) of medical therapy indicates need for elective surgery even if initial biopsy was benign — due to the risk of sampling error missing an underlying malignancy [2][3].
For patients who receive endoscopic therapy for Forrest Class Ia, Ib, IIa, or IIb ulcer bleeding:
IV esomeprazole (or pantoprazole) 80 mg stat bolus → 8 mg/hour continuous infusion for 72 hours [3]
Why this protocol?
- Platelet aggregation and clot stability are pH-dependent — platelet function is optimal at pH > 6.0. Below pH 5.4, platelet aggregation is abolished; below pH 4, fibrin clots dissolve (pepsin-mediated fibrinolysis).
- High-dose IV PPI maintains intragastric pH > 6 for a sustained period → stabilises the haemostatic clot over the bleeding vessel → reduces rebleeding.
- After 72 hours, switch to oral PPI (e.g., esomeprazole 40 mg daily).
Two major scores are used to stratify patients with UGIB:
| Parameter | Glasgow-Blatchford Score (GBS) | Rockall Score |
|---|---|---|
| When used | Pre-endoscopy (does NOT require OGD findings) | Post-endoscopy (includes OGD findings) |
| Components | Clinical + Lab: Hb, urea, BP, pulse, melaena, syncope, liver disease, heart failure | Clinical (Age, BP, Comorbidities) + Endoscopic (Diagnosis, Evidence of bleeding) — "ABCDE" [5] |
| Purpose | Identifies patients who do NOT need intervention (GBS = 0 → very low risk, can be discharged with outpatient OGD) | Predicts mortality after UGIB |
| Key threshold | GBS = 0 → safe for outpatient management | Higher score → higher mortality |
| Test | Indication | Key Findings |
|---|---|---|
| Fasting serum gastrin level | Suspected Zollinger-Ellison syndrome (recurrent/refractory ulcers, unusual location, H. pylori-negative, NSAID-negative) | Markedly elevated gastrin ( > 1000 pg/mL) in the presence of high gastric acid output (to exclude PPI-induced or achlorhydria-induced hypergastrinaemia) [2] |
| Secretin stimulation test | Confirm ZES when gastrin is equivocal (100–1000 pg/mL) | IV secretin → paradoxical ↑ gastrin by > 120 pg/mL in ZES (normal: gastrin stays the same or decreases) |
| Gastric acid output analysis (BAO/MAO) | Suspected ZES or acid hypersecretory state | Basal acid output (BAO) > 15 mEq/h (or > 5 mEq/h post-surgery) suggests ZES |
| CT/MRI abdomen | Localise gastrinoma if ZES confirmed | Duodenal/pancreatic mass in the "gastrinoma triangle" (confluence of cystic duct, D2/D3 junction, and neck/body of pancreas) |
| Somatostatin receptor scintigraphy (Octreoscan) or ⁶⁸Ga-DOTATATE PET/CT | Localise gastrinoma (most sensitive) | Somatostatin receptor-positive tumour uptake |
| Serum calcium, PTH | Screen for MEN1 in ZES patients | ↑ Ca²⁺ + ↑ PTH → primary hyperparathyroidism as part of MEN1 |
| Scenario | First-Line Investigations | Key Investigation | What You're Looking For |
|---|---|---|---|
| Uncomplicated dyspepsia, no alarm features, age < 55 | Non-invasive H. pylori test (UBT or SAT) | UBT | Active H. pylori infection → eradicate |
| Dyspepsia WITH alarm features or age ≥ 55 | OGD + biopsy | OGD | Ulcer morphology (benign vs malignant), H. pylori status, other pathology |
| Acute UGIB | CBC, clotting, X-match, LRFT, VBG [5][8] → urgent OGD | OGD within 24h | Forrest classification → guide therapy; identify source |
| Suspected perforation | Erect CXR [2][5][8] → CT if CXR negative | Erect CXR (free gas) | Pneumoperitoneum; if positive + peritonism → surgery |
| Suspected GOO | ABG + electrolytes [3] → AXR → OGD after decompression | ABG: hypoCl hypoK metabolic alkalosis; OGD: biopsy to exclude malignancy | Metabolic derangement; benign vs malignant obstruction |
| Refractory/recurrent PUD, H. pylori-negative, NSAID-negative | Fasting serum gastrin + gastric acid output | Fasting serum gastrin | ZES (gastrinoma); MEN1 screen |
High Yield Summary
- OGD is the gold standard for diagnosing PUD — both diagnostic and therapeutic [2][9].
- Alarm features → urgent OGD. No alarm features and age < 55 → "test and treat" with non-invasive H. pylori testing [2].
- ALL patients with endoscopy-diagnosed PUD must be tested for H. pylori — CLO test (1 biopsy) + histology (2 biopsies) from the antrum (+ body to increase sensitivity) [2].
- Stop PPIs ≥ 2 weeks and antibiotics ≥ 4 weeks before H. pylori testing to avoid false negatives.
- UBT is the best non-invasive test for both diagnosis and confirmation of eradication [2].
- Serology cannot distinguish active from past infection — do NOT use it to confirm eradication.
- Forrest classification guides endoscopic therapy: Class I and IIa/IIb → endoscopic dual therapy + IV PPI infusion. Class IIc and III → PPI alone, early discharge [1][2].
- Dual endoscopic therapy: adrenaline injection + heater probe/clip. Adrenaline alone is insufficient [1][3].
- Post-OGD IV PPI infusion (80 mg bolus → 8 mg/h × 72h) — stabilises clot by maintaining pH > 6. For ulcer bleeding ONLY, not varices [3].
- All gastric ulcers must be biopsied (multiple from rim). Follow-up OGD mandatory until healing confirmed. Non-healing GU > 12 weeks → surgery even if biopsy benign [2][3].
- Erect CXR for suspected PPU — sensitivity ~75–80%; if negative but clinical suspicion persists → CT [2][5].
- Elevated urea:creatinine ratio > 100:1 is a clue to UGIB [5].
- Glasgow-Blatchford Score: pre-endoscopy; GBS = 0 → safe outpatient management. Rockall Score: post-endoscopy; predicts mortality [5].
- Avoid endoscopy in suspected perforation — gas insufflation can convert a sealed-off perforation to free perforation [5].
Active Recall - PUD Diagnostic Criteria, Algorithm & Investigations
References
[1] Lecture slides: GC 198. Profuse vomiting of fresh blood and in shock severe upper GI bleeding.pdf (p18 — ulcer bleed stops spontaneously 70–80%; p19 — general guideline algorithm; p24 — bleeding peptic ulcer: clean base, therapeutic endoscopy, PPI; p29 — choice of additional procedure) [2] Senior notes: felixlai.md (PUD sections pp. 386–391 — overview, classification, etiology, diagnosis, H. pylori testing, OGD findings, Forrest classification, follow-up endoscopy; Dyspepsia sections pp. 327–330 — alarm features, test-and-treat algorithm) [3] Senior notes: maxim.md (Benign diseases of stomach pp. 127–131 — PUD classification, surgical management, GOO, perforation post-op management; UGIB therapeutic endoscopy p. 53 — dual therapy, adrenaline, heater probe, haemospray, clip, PPI infusion, Forrest classification) [5] Senior notes: maxim.md (UGIB pp. 52–53 — pre-endoscopy management, bloods, risk stratification GBS and Rockall; Acute abdomen pp. 43–45 — investigations, imaging, avoid endoscopy for acute abdomen) [8] Lecture slides: GC 195. Lower and diffuse abdominal pain RLQ problems; pelvic inflammatory disease; peritonitis and abdominal emergencies.pdf (p12 — investigations: bedside tests, blood tests, imaging, endoscopy) [9] Senior notes: felixlai.md (OGD indications and contraindications pp. 76–77)
Management of Peptic Ulcer Disease — Algorithm & Treatment Modalities
The management of PUD is best understood as a series of concentric layers, moving from the most common scenario (uncomplicated ulcer treated medically) outward to the rarest (surgical emergency). At every step, the question is: What is the aetiology, and what is the complication? — because treatment is entirely driven by these two answers.
2. Management of Uncomplicated PUD
The three pillars of uncomplicated PUD management are:
- Remove the cause (H. pylori eradication, NSAID withdrawal)
- Heal the ulcer (acid suppression with PPI)
- Prevent recurrence (confirm eradication, prophylaxis if NSAIDs cannot be stopped)
A. Medication Alteration
-
NSAID users [2]:
- Switch to less ulcerogenic NSAIDs or COX-2 selective inhibitors — COX-2 inhibitors (e.g., celecoxib) spare COX-1-mediated prostaglandin synthesis in the gastric mucosa, preserving the mucosal barrier. Why does this work? Because the ulcer is caused by systemic COX-1 inhibition reducing PGE₂; removing the offending agent allows prostaglandin levels to recover.
- Withdraw NSAIDs during PPI treatment — healing is dramatically impaired if the causative agent continues [2].
-
Aspirin users [2]:
- Bleeding peptic ulcer: Resume aspirin with PPI treatment once haemostasis is secured — the rationale is to minimise cardiovascular risk. Studies show that withholding aspirin for > 7 days after ulcer haemostasis is associated with increased cardiovascular events (MI, stroke) without significant benefit in reducing rebleeding [2][3].
- Non-bleeding peptic ulcer: Continue aspirin with PPI treatment — no need to stop aspirin if the ulcer is not actively bleeding [2].
-
Antithrombotic management principles [3]:
- Withhold all antithrombotics stat +/- reversal agents in acute bleeding
- Resume aspirin after OGD; clopidogrel 5–7 days later [3] — aspirin is resumed early because its cardiovascular benefit outweighs the small rebleeding risk once endoscopic haemostasis is achieved.
Aspirin in Bleeding PUD — Don't Be Afraid to Restart
A common mistake is to permanently stop aspirin after a bleeding ulcer. This increases mortality from cardiovascular events. The correct approach is: achieve haemostasis → restart aspirin with PPI cover as soon as possible (typically within 3–7 days). The PPI provides the ulcer protection while aspirin provides the cardiovascular protection [2][3].
B. Lifestyle Modification [2]
- Smoking cessation — smoking impairs mucosal blood flow, reduces bicarbonate secretion, promotes duodenogastric reflux, and delays ulcer healing. It also increases recurrence rates even after successful H. pylori eradication.
- Limit alcohol intake — alcohol is a direct mucosal irritant and stimulates acid secretion.
2.3 Medical Treatment — Acid Suppression
PPIs are the backbone of PUD treatment. Understanding their mechanism from first principles:
Mechanism: PPIs are prodrugs that are absorbed in the small intestine, enter the systemic circulation, and accumulate in the acidic canaliculi of parietal cells (the only cells with pH < 2 in the body). In this acidic environment, they are converted to their active form (a sulphenamide), which irreversibly binds to and inhibits the H⁺/K⁺-ATPase (proton pump) — the final common pathway of acid secretion, regardless of what stimulus triggered it (gastrin, histamine, or acetylcholine).
- "Proton pump" → the H⁺/K⁺-ATPase that pumps H⁺ into the gastric lumen in exchange for K⁺
- "Inhibitor" → irreversible covalent binding → acid secretion only returns when new proton pumps are synthesised (~24–48 hours)
Why PPIs are superior to H₂ blockers: H₂ receptor antagonists (e.g., ranitidine, famotidine) only block the histamine-mediated pathway of acid secretion. PPIs block the final common pathway regardless of stimulus → more complete and prolonged acid suppression.
| PPI | Dose for Ulcer Healing | Notes |
|---|---|---|
| Omeprazole | 20 mg OD | First PPI developed |
| Esomeprazole | 20–40 mg OD | S-isomer of omeprazole; marginally better bioavailability |
| Pantoprazole | 40 mg OD | Available IV — used for IV PPI infusion (80 mg stat → 8 mg/h × 72h) [1][3] |
| Lansoprazole | 30 mg OD | — |
| Rabeprazole | 20 mg OD | Fastest onset; less dependent on CYP2C19 metabolism |
Duration of PPI therapy:
- DU: 4 weeks (8 weeks if large or complicated)
- GU: 8 weeks (because GU heal more slowly; also need to confirm healing on follow-up OGD)
- NSAID-related ulcers: 8 weeks (or longer if NSAID cannot be stopped)
PPI side effects (important for long-term use):
- ↑ Risk of Clostridioides difficile infection (acid suppression allows gut colonisation)
- ↑ Risk of community-acquired pneumonia (acid suppression allows oropharyngeal bacterial overgrowth → aspiration)
- ↓ Calcium and magnesium absorption → osteoporosis risk with long-term use
- Vitamin B12 deficiency (acid needed for B12 release from food proteins)
- Theoretical CYP2C19 interaction with clopidogrel (mainly omeprazole) — consider pantoprazole instead
- Mechanism: Competitive, reversible blockade of H₂ histamine receptors on parietal cells → ↓ histamine-stimulated acid secretion
- H₂ blocker and PPI hasten healing of ulcers [1]
- Less effective than PPIs; now used mainly as adjunct or when PPIs are contraindicated
- Examples: famotidine (preferred since ranitidine was withdrawn due to NDMA contamination)
| Agent | Mechanism | Use in PUD |
|---|---|---|
| Misoprostol | Synthetic PGE₁ analogue → replaces NSAID-depleted prostaglandins → ↑ mucus, ↑ bicarbonate, ↑ mucosal blood flow, ↓ acid secretion | Prophylaxis of NSAID-induced ulcers. Contraindicated in pregnancy (uterotonic → can cause abortion) |
| Sucralfate | Polymerises in acidic pH → forms a viscous gel that binds to ulcer base → physical barrier against acid/pepsin | Rarely used now; useful in stress ulcer prophylaxis in ICU |
| Bismuth subsalicylate/subcitrate | Coats ulcer base + direct bactericidal activity against H. pylori + stimulates PG/bicarbonate secretion | Component of bismuth quadruple therapy |
| Antacids (aluminium/magnesium hydroxide) | Directly neutralise gastric acid | Symptomatic relief only; do NOT heal ulcers; Al causes constipation, Mg causes diarrhoea |
2.4 Medical Treatment — H. pylori Eradication
This is the single most important treatment for H. pylori-positive PUD. Successful eradication reduces ulcer recurrence from ~80% to < 5% per year.
A. Standard PPI-Based Triple Therapy (14 days)
| Component | Drug | Dose | Rationale |
|---|---|---|---|
| PPI | Omeprazole / Esomeprazole / etc. | Standard dose BD | ↑ Intragastric pH → improves antibiotic stability and efficacy (clarithromycin is degraded at low pH); ↑ H. pylori growth rate (replicating bacteria are more susceptible to antibiotics) |
| Antibiotic 1 | Amoxicillin | 1 g BD | Bactericidal; disrupts cell wall synthesis. Low resistance rates |
| Antibiotic 2 | Clarithromycin | 500 mg BD | Bacteriostatic (inhibits 50S ribosomal subunit → protein synthesis). Most potent anti-H. pylori antibiotic, but resistance is rising (20–30% in Hong Kong) |
B. Bismuth Quadruple Therapy (14 days) — preferred if clarithromycin resistance > 15% or as second-line
| Component | Drug | Dose | Rationale |
|---|---|---|---|
| PPI | Standard dose BD | — | Acid suppression |
| Bismuth | Bismuth subcitrate / subsalicylate | QDS | Direct bactericidal; coats ulcer; disrupts H. pylori cell membrane |
| Metronidazole | 400 mg TDS–QDS | Anaerobe-active; creates toxic free radicals within H. pylori | |
| Tetracycline | 500 mg QDS | Inhibits 30S ribosome → protein synthesis; broad spectrum |
Choosing First-Line Therapy in Hong Kong
In Hong Kong, clarithromycin resistance has risen to ~20–30%. Current guidelines therefore increasingly favour bismuth quadruple therapy as first-line, or recommend susceptibility-guided therapy where culture results are available. If clarithromycin resistance is < 15% in the local population, standard triple therapy remains acceptable. Always check local resistance data [2].
Levofloxacin-based Triple Therapy (14 days):
- PPI (standard dose BD) + Amoxicillin (1 g BD) + Levofloxacin (500 mg OD)
- Levofloxacin is a fluoroquinolone with high activity against Gram-negative bacilli including H. pylori, with lower resistance rates than clarithromycin in many populations
Or: If first-line was triple therapy → use bismuth quadruple therapy as second-line (and vice versa).
- Bacterial culture with antibiotic sensitivity testing should be performed [2] — this is the only indication for culture in routine PUD management
- Susceptibility-guided therapy based on resistance patterns
- Consider rifabutin-based regimens (rifabutin + amoxicillin + PPI) as a last resort
- Urea breath test (UBT) is the preferred test for confirming eradication
- Performed ≥ 4 weeks after completing eradication therapy AND ≥ 2 weeks after stopping PPI
- Stool antigen test is an acceptable alternative
- Serology is NOT valid for confirming eradication (antibodies persist for months)
This is a high-yield topic — the prevention strategy depends entirely on the underlying cause:
| Aetiology | Prevention Strategy | Key Points |
|---|---|---|
| H. pylori ulcers | H. pylori eradication | Maintenance acid suppression with PPI is NOT necessary after successful eradication [2] — once the cause is removed, the ulcer does not recur |
| NSAID ulcers | Review need for NSAIDs; avoid in patients with high GI risk or prior complicated PUD; add PPI or misoprostol as prophylaxis with NSAIDs or switch to COX-2 inhibitor [2] | High GI risk defined as ≥ 2 of: age > 65, previous PUD, high-dose NSAIDs, concomitant aspirin/corticosteroids/anticoagulants [2] |
| Aspirin ulcers | Review need for aspirin; add PPI as prophylaxis with aspirin use [2] | PPI co-prescription is standard for all patients on aspirin with ANY additional GI risk factor |
| Idiopathic ulcers | Maintenance acid suppression therapy with PPI long-term [2] | Since no removable cause is identified, continuous PPI is needed to prevent recurrence |
H. pylori Ulcers Do NOT Need Maintenance PPI
After successful H. pylori eradication, maintenance PPI is unnecessary [2]. The ulcer recurrence rate drops to < 5%/year. This distinguishes H. pylori ulcers from NSAID and idiopathic ulcers, where ongoing acid suppression is needed.
3. Management of Complicated PUD
3.1 Haemorrhage — The Most Common and Deadly Complication
Haemorrhage is the leading cause of death from peptic ulcer [3].
This follows the ABC approach — treat the patient, not the endoscopy report:
- Airway: Cuffed endotracheal (ET) tube + NG tube if massive haematemesis or impaired consciousness — to prevent aspiration [5]
- Breathing: O₂ supplementation [5]
- Circulation [5]:
- NPO
- 2 large-bore IV cannulae (at least 16G, preferably 14G — in the antecubital fossae)
- IV normal saline 2L fast run to maintain BP/pulse + urine output
- Blood transfusion: Hb < 7 g/dL in low-risk patients; Hb < 9 g/dL in high-risk patients (e.g. elderly, CAD) [5]
- Closely monitor pulse, BP, I/O [5]
- Withhold anticoagulants and antiplatelets (balance thrombotic risk when considering reversal) [3][5]
- FFP for coagulopathy; platelets for thrombocytopenia [5]
Ulcer bleed stops spontaneously in approximately 70–80% of cases [1]. The key is to identify patients in shock and those with ongoing bleeding [1].
- Bloods: CBC, clotting, cross-match, LRFT, VBG
- Pre-endoscopic PPI: IV esomeprazole 80 mg stat → 8 mg/h infusion until OGD — however, HO handbook states this is only if early endoscopy cannot be arranged [5]. The rationale is that raising intragastric pH stabilises any forming clot, potentially downgrading the Forrest class before endoscopy.
- Risk stratification: Glasgow-Blatchford Score (pre-endoscopy) / Rockall Score (post-endoscopy) [5]
- Arrange early upper endoscopy within 24 hours after initial stabilisation [5]
Acute treatment depends on diagnosis → bleeding peptic ulcer [1]:
Forrest Class determines management:
| Risk | Forrest Class | Management |
|---|---|---|
| High risk | Ia (spurting), Ib (oozing), IIa (visible vessel), IIb (adherent clot) | Endoscopic dual therapy + IV PPI infusion |
| Low risk | IIc (flat pigmented spot), III (clean base) | Oral PPI; clean base → start feeding, early discharge [1] |
Therapeutic endoscopy modalities [1][3]:
| Method | Modality | Mechanism |
|---|---|---|
| Injection method | Adrenaline 1:10,000 [1][3] | Tamponade (volume effect compresses vessel) + vasoconstriction (α₁-adrenergic) + platelet aggregation (promotes haemostasis). Must NOT be used alone — only provides temporary haemostasis; must combine with thermal or mechanical method |
| Thermal method | Heater probe [1][3] | Coaptive effect: firm pressure applied to the vessel wall + heat delivered simultaneously → coagulates and seals the vessel. The pressure component is key — it apposes the vessel walls before the heat fuses them |
| Mechanical method | Metal clip [1][3] | Physically grasps and occludes the bleeding vessel. More prolonged action than thermal methods. Particularly good for discrete visible vessels |
| Mechanical method | Haemospray [3] | Inorganic powder that acts as a mechanical barrier + absorbent (absorbs water from blood → concentrates clotting factors → promotes haemostasis) — the "雲南白藥 principle." Used for large-area oozing where clips or heater probe are impractical |
Dual therapy (adrenaline injection + heater probe or clip) is the standard — injection therapy should NOT be used as monotherapy since it is associated with a high rate of recurrent bleeding compared to other modalities [2][3].
- Post-OGD PPI infusion: pantoprazole/esomeprazole 80 mg stat, then 8 mg/h for 72 hours [3]
- Indicated for endoscopic treatment of Forrest Class Ia, Ib, IIa, IIb ulcer bleeding [3]
- Rationale: higher pH stabilises clot since platelet function depends on normal pH — platelet aggregation is abolished at pH < 5.4; fibrin clots dissolve at pH < 4 (pepsin-mediated fibrinolysis) [3]
- NOT efficacious for variceal bleeding — only give oral PPI to reduce post-banding ulcers [3]
- Post-endoscopic monitoring of Hb count to identify rebleeding [2]
- Signs of rebleeding: haematemesis, fresh melaena, tachycardia, falling Hb trend, blood in NG tube [2]
- After stabilisation: H. pylori eradication + review NSAID use
Indications for surgery for bleeding ulcer [1][2]:
- Therapeutic endoscopist not available [1]
- Massive bleeding (cannot visualise bleeding point) [1]
- Failed endoscopic therapy [1]
- Rebleed after endoscopic therapy [1]
- Haemodynamic instability despite vigorous fluid resuscitation [2]
- Continuous slow bleeding with transfusion > 3 units per day [2]
Surgery = Plication of bleeder + additional procedure [1]
The "plication of bleeder" means suture ligation of the bleeding vessel (e.g., GDA in posterior DU). The "additional procedure" depends on the ulcer type and surgeon's judgment:
| Type of ulcer | Choice of additional procedure [1] |
|---|---|
| DU | V + P (truncal Vagotomy + Pyloroplasty) [1] — vagotomy reduces acid secretion; pyloroplasty is needed because vagotomy paralyses the pylorus (loss of vagal-mediated pyloric relaxation) → without pyloroplasty, the stomach cannot empty |
| GU | Partial gastrectomy [1] — because any gastric ulcer may harbour malignancy, the ulcer must be resected (not just over-sewn) [3] |
Factors determining the choice of additional procedure [1]:
- Condition of patient (haemodynamic stability, comorbidities)
- Experience of surgeon
- Type of ulcer (DU vs GU)
Transcatheter Arterial Embolisation (TAE) [2][3]:
- Alternative to surgery when the patient is unfit for surgery or as a bridge
- Interventional radiology performs angiography of the coeliac trunk and SMA → identifies contrast extravasation → selective cannulation of the bleeding vessel → angiographic coiling from distal to proximal until extravasation ceases [2]
- Equally effective as surgery in patients who failed therapeutic endoscopy and associated with fewer complications [2]
- Reduces need for surgery without increasing overall mortality [2]
- Limitations of endoscopic haemostasis that trigger surgical/TAE referral [3]:
- Massive bleeding obscuring view
- Large bleeding artery ( > 3.2 mm)
- Large ulcer ( > 2 cm) [3]
Most common site: anterior wall of D1 [3].
Management Algorithm [3]
Step 1: Resuscitation
- NPO
- IV fluids
- NG tube decompression (may have co-existing GOO) [3]
- IV broad-spectrum antibiotics (e.g., augmentin) — to cover the bacterial peritonitis that develops within 6 hours [3]
- IV PPI / H₂ blocker (suppress acid production, limit ongoing chemical peritonitis) [3]
Step 2: Surgery [3]
Treatment options: H. pylori eradication + surgery + peritoneal lavage ( > 6L NS) after repair [3]
| Ulcer Size/Type | Surgical Approach |
|---|---|
| Small DU or GU ( < 2 cm) | Laparoscopic or open (Graham) omental patch repair [3]. The omentum is a "biological plug" — a pedicled flap of omentum is sewn over the perforation to seal it. Alternatives if omentum is not available: falciform ligament, jejunal serosal patch [3] |
| Large DU | Distal gastrectomy + Billroth II reconstruction +/- controlled duodenostomy (especially if difficult duodenal stump) [3] |
| Large GU | Wedge excision or partial gastrectomy + Billroth II reconstruction [3] — must resect because any GU could be malignant |
Biopsy ulcer edge for ALL gastric ulcers to rule out malignancy [3]
Indications to convert laparoscopic to open [3]:
- Haemodynamically unstable → shorter operative time with open
- Large defect ( > 1 cm) → difficult laparoscopic suturing
- Gross contamination → limited laparoscopic peritoneal lavage capacity
Post-operative management [3]:
- NPO until no signs of leakage and return of bowel function
- Complete full course of antibiotics
- Continue PPI until follow-up OGD
- FU OGD 6–8 weeks after surgery to confirm healing and check H. pylori status [3]
Prognosis: Boey's Score (3 points) [3]:
| Risk Factor | Points |
|---|---|
| Time from perforation to admission > 24 hours | 1 |
| Pre-op SBP < 100 mmHg | 1 |
| ≥ 1 systemic illness (heart disease, liver disease, renal disease, DM) | 1 |
| Mortality: Score 0 = 0%, Score 1 = 10%, Score 2 = 45.5%, Score 3 = 100% |
GOO results from chronic scarring and fibrosis at the pylorus/D1 from repeated ulcer-healing cycles, or from acute oedema and spasm during active ulceration. Prolonged obstruction can lead to gastric atony (the dilated stomach loses its contractile ability) [2].
GOO is malignant until proven otherwise — 80% malignant (gastric cancer is the most common cause), 20% benign (PUD-related pyloric stenosis is the 2nd most common overall cause) [3].
Management [2][3]
Step 1: Medical ("Drip and Suck") [2][3]
- NPO
- Nasogastric (NG) tube — decompress the stomach, relieve vomiting
- Fluid resuscitation
- Correction of electrolyte abnormality [2]:
- Normal saline + KCl — prolonged vomiting causes hypochloraemic hypokalaemic metabolic alkalosis [2]
- Why NS specifically? Because this is a chloride-responsive alkalosis — the kidneys need chloride to excrete excess bicarbonate; until Cl⁻ is replaced, the alkalosis cannot correct
- IV PPI — antisecretory agents remain the mainstay of initial treatment (e.g., pantoprazole) [2]
Step 2: OGD after decompression [3]
- Once the stomach is adequately decompressed → OGD to biopsy the pylorus and establish whether GOO is benign (PUD) or malignant [3]
Step 3: Definitive Treatment [2][3]
| Cause | Treatment |
|---|---|
| Benign (PUD-related) | Endoscopic balloon dilatation [2] +/- duodenal stenting; if fails or recurs → surgery |
| Malignant | Curative: treat underlying CA; Palliative: surgical bypass (gastrojejunostomy) or endoscopic self-expandable metal stent (SEMS) [3] |
Surgical options for benign GOO [2][3]:
- GU: Antrectomy with Billroth I/II reconstruction [2]
- DU: Truncal vagotomy + Antrectomy with Billroth I/II reconstruction [2]
- Alternative: Bypass (gastrojejunostomy), pyloroplasty [3]
- Penetration = ulcer erodes through the bowel wall without free perforation — the adjacent organ "seals" the defect, preventing spillage into the peritoneal cavity [2]
- Occurs in descending order of frequency: Pancreas > Lesser omentum > Biliary tract > Liver > Greater omentum > Mesocolon > Colon > Vascular structures [2]
- Leads to complications such as pancreatitis (most common — posterior DU penetrating into pancreas) or cholangitis
- Clinical clue: pain becomes more intense, longer-lasting, localised to the back, and is NOT relieved by food or antacids (unlike uncomplicated DU) [2]
- Surgical treatment is NOT recommended [2] — manage conservatively with IV PPI and treat the secondary complication (e.g., pancreatitis). Surgery is only considered if the penetration leads to uncontrollable haemorrhage or fistula formation.
Surgery is rarely required now [3] due to effective medical therapy, but indications persist:
| Indication | Rationale |
|---|---|
| Complicated PUD (haemorrhage, perforation, GOO) | Emergency or semi-elective depending on clinical scenario [2][3] |
| Refractory to medical treatment | Zollinger-Ellison syndrome (gastrinoma) should be excluded before performing elective surgery for PUD [2] — ZES causes refractory ulcers but is treated medically (high-dose PPI) or by tumour resection, not by acid-reducing surgery |
| Suspicious of malignancy | Non-healing GU after 12 weeks of medical therapy indicates need for elective surgery even if biopsy is benign [2] — due to sampling error risk |
| Ulcer Type | Surgical Options | Principle |
|---|---|---|
| DU (acid reduction) | Highly selective vagotomy (HSV): nerve of Latarjet preserved, but technically difficult | Denervate parietal cell mass only; preserve antral motility → no drainage needed |
| Truncal vagotomy + drainage (pyloroplasty or gastrojejunostomy): nerve of Latarjet sacrificed | Denervate entire stomach → pyloric dysfunction → must add drainage procedure | |
| Gastrectomy (antrectomy / distal gastrectomy) + reconstruction (Billroth II > Roux-en-Y) | Remove antral G cells (↓ gastrin) + acid-secreting mucosa | |
| GU (prevent malignancy) | Type I: distal gastrectomy + Billroth II | Resect ulcer to exclude/treat possible malignancy |
| Type II / III: truncal vagotomy + antrectomy + Billroth II | Acid-hypersecretory types — need acid reduction + ulcer resection | |
| Type IV: subtotal gastrectomy (extending to ulcer) + Billroth I / II / Roux-en-Y | High lesser curvature ulcer near GOJ — technically challenging, requires extensive resection |
Reconstruction Types (brief explanation):
- Billroth I (gastroduodenostomy): stomach remnant anastomosed directly to duodenum — most physiological, but limited by tension if too much stomach is resected
- Billroth II (gastrojejunostomy): stomach remnant anastomosed to jejunum (side-to-side) — more versatile, less anastomotic tension; preferred for most PUD surgery [3]
- Roux-en-Y: Y-shaped jejunal limb; bile is diverted away from the gastric remnant → reduces bile reflux gastritis. Used when bile reflux is a concern (e.g., Type IV GU, revision surgery)
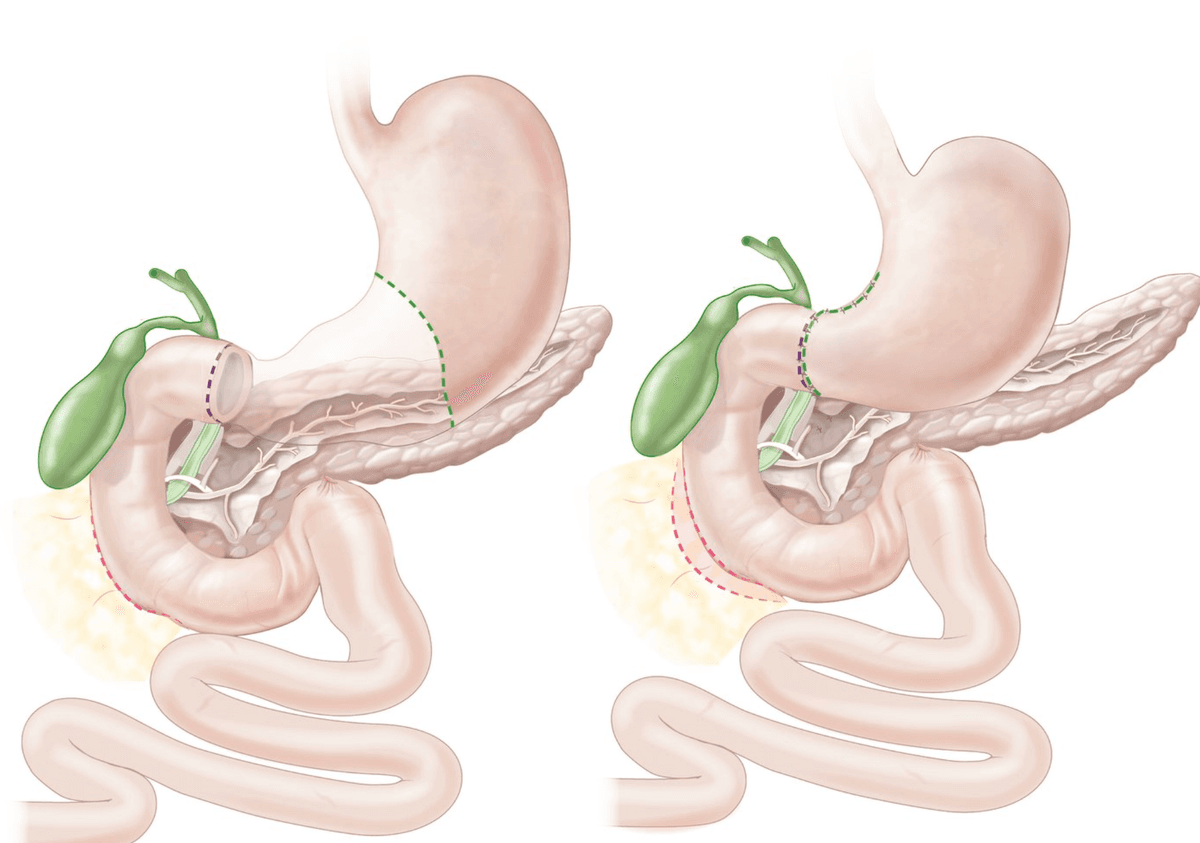
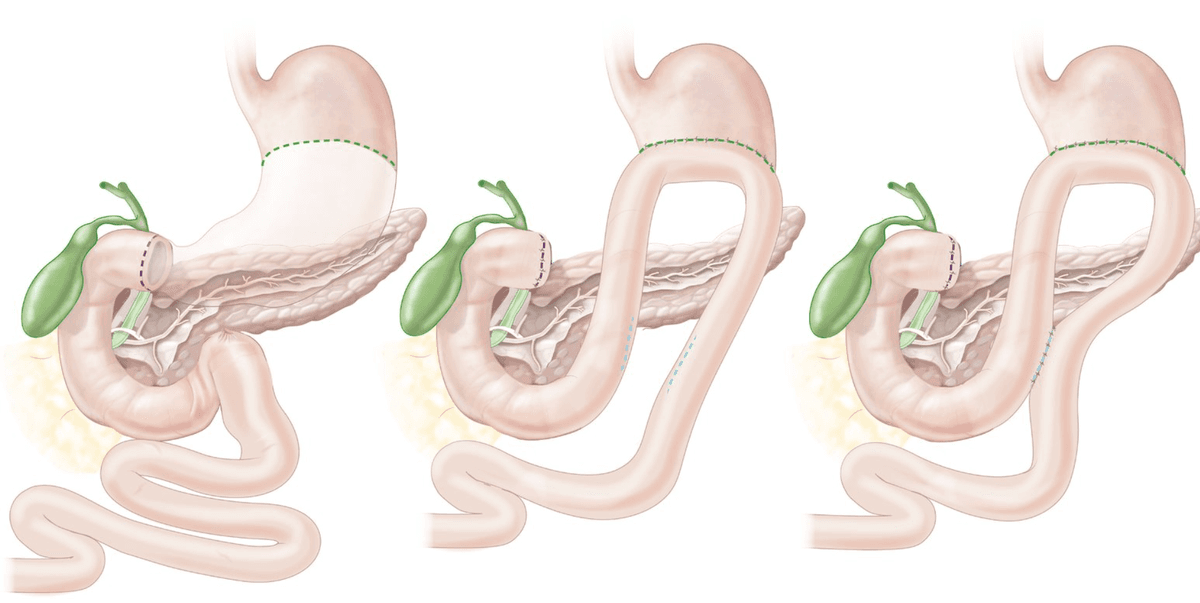
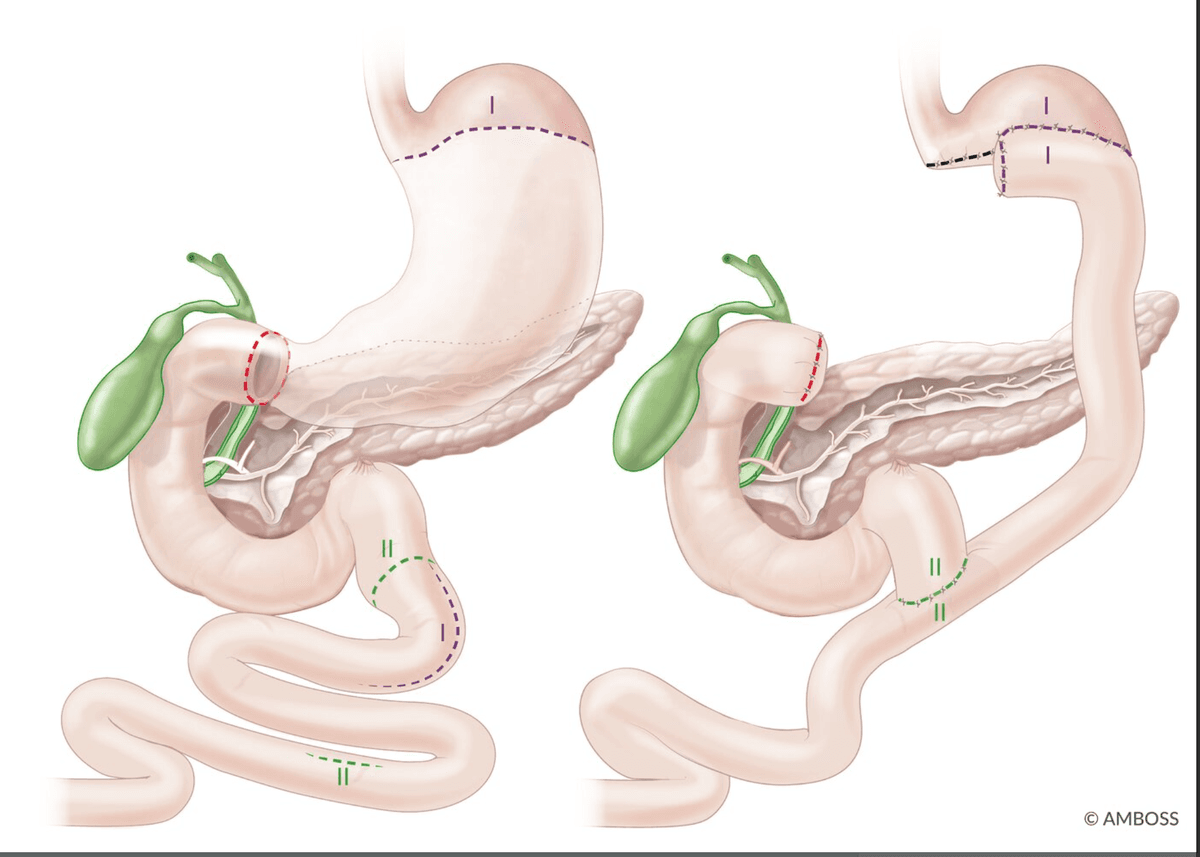
Understanding these complications is essential because they explain why surgery is a last resort:
| Syndrome | Mechanism | Features |
|---|---|---|
| Dumping syndrome (early) | Rapid emptying of hyperosmolar gastric contents into small bowel → fluid shifts into the lumen → hypovolaemia + bowel distension | 15–30 min after meals: nausea, cramping, diarrhoea, dizziness, diaphoresis, tachycardia |
| Dumping syndrome (late) | Rapid glucose absorption → exaggerated insulin release → reactive hypoglycaemia | 1–3 hours after meals: tremor, diaphoresis, confusion |
| Bile reflux gastritis (alkaline reflux) | Loss of pyloric sphincter → bile refluxes into gastric remnant → chronic gastritis | Burning epigastric pain, bilious vomiting, NOT relieved by antacids. Roux-en-Y revision may be needed |
| Afferent loop syndrome (Billroth II) | Obstruction/kinking of the afferent limb → bile/pancreatic juice accumulates → sudden decompression | Sudden bilious vomiting that relieves pain; abdominal distension |
| Nutritional deficiencies | Loss of absorptive surface + loss of intrinsic factor (parietal cells) + reduced mixing | Iron deficiency (reduced acid → reduced Fe²⁺ absorption), B12 deficiency, calcium/vitamin D malabsorption → osteoporosis |
| Marginal (stomal) ulcer | Ulcer at the surgical anastomotic site | Recurrent epigastric pain; check for retained antrum, incomplete vagotomy, or H. pylori |
High Yield Summary
- Uncomplicated PUD management = remove the cause + heal the ulcer + prevent recurrence.
- H. pylori eradication is the most important treatment for H. pylori-positive PUD. Standard triple therapy (PPI + amoxicillin + clarithromycin × 14 days) or bismuth quadruple therapy. Confirm eradication with UBT ≥ 4 weeks post-Rx.
- H. pylori ulcers do NOT need maintenance PPI after successful eradication [2]. NSAID and idiopathic ulcers DO.
- Aspirin in bleeding PUD: resume with PPI cover once haemostasis secured — do NOT permanently stop [2][3].
- Ulcer bleed stops spontaneously in ~70–80% [1]. Identify shock and ongoing bleeding.
- Endoscopic dual therapy (adrenaline + heater probe/clip) for Forrest Ia, Ib, IIa, IIb. Injection therapy alone is NOT sufficient [2][3].
- Post-OGD IV PPI infusion (80 mg stat → 8 mg/h × 72h) for high-risk ulcer bleeding. NOT for varices [3].
- Clean base (Forrest III): start feeding, early discharge [1].
- Surgery for bleeding ulcer: plication of bleeder + additional procedure. DU: V+P; GU: partial gastrectomy [1].
- TAE is an alternative to surgery if patient is unfit; equally effective with fewer complications [2].
- Perforation: Small — omental patch; Large DU — gastrectomy + Billroth II; Large GU — wedge excision/gastrectomy. Always biopsy GU edges [3].
- GOO: Drip and suck → OGD + biopsy → malignant until proven otherwise [3].
- Penetration: surgery NOT recommended; treat conservatively [2].
- Elective surgery indications: complicated PUD, refractory to medical Rx (exclude ZES first), non-healing GU > 12 weeks [2][3].
- Boey's Score for PPU prognosis: delay > 24h, SBP < 100, systemic illness (0–3 points) [3].
Active Recall - PUD Management
References
[1] Lecture slides: GC 198. Profuse vomiting of fresh blood and in shock severe upper GI bleeding.pdf (p18 — ulcer bleed stops spontaneously 70–80%; p19 — general guideline algorithm; p23 — acute treatment depends on diagnosis; p24 — bleeding peptic ulcer: clean base, therapeutic endoscopy, PPI infusion; p28 — surgery for bleeding ulcer: indications; p29 — choice of additional procedure: DU V+P, GU partial gastrectomy) [2] Senior notes: felixlai.md (PUD treatment pp. 391–398 — indications for surgery, supportive treatment, medical treatment, H. pylori eradication, complications management, prevention; UGIB treatment pp. 340–341 — general management, medications, surgery) [3] Senior notes: maxim.md (Benign diseases of stomach — PUD surgical management, PUD complications pp. 127–131; UGIB therapeutic endoscopy — dual therapy, PPI infusion, antithrombotic management p. 53; Perforation — Boey's score, post-op management pp. 128–130) [5] Senior notes: maxim.md (UGIB initial management — resuscitation, pre-endoscopy management, risk stratification pp. 52–53)
Complications of Peptic Ulcer Disease
PUD is not simply "a sore that hurts." Left untreated or in the setting of ongoing risk factors, the ulcer can breach deeper layers of the bowel wall and injure surrounding structures. Every complication is best understood by asking: which layer has the ulcer just breached, and what lies immediately beyond it?
The five major complications, in order of clinical importance:
- Haemorrhage (most common complication; leading cause of death [3])
- Perforation (most dramatic acute presentation)
- Gastric outlet obstruction (GOO) (chronic complication)
- Penetration (contained perforation into an adjacent organ)
- Fistulisation (rarest)
Additionally, there is a sixth category — malignant transformation — which applies to gastric ulcers only, and a seventh — post-surgical (gastrectomy) complications — which is crucial for exams.
1. Haemorrhage
The ulcer starts at the mucosa and erodes progressively deeper: mucosa → muscularis mucosae → submucosa → muscularis propria → serosa. The submucosa contains a rich vascular plexus. Once the ulcer breaches the muscularis mucosae and enters the submucosa, it encounters arteries and veins. If the ulcer erodes even deeper through the full wall thickness posteriorly, it can reach named arteries behind the bowel wall.
- DU bleeding usually locates at the posterior duodenal wall within 2 cm of the pylorus and typically erodes into the gastroduodenal artery (GDA) [2][3] — the GDA runs behind D1, so a posterior DU bores straight into it.
- GU bleeding on the lesser curvature can erode into the left gastric artery (the largest arterial supply to the stomach).
- The clinical presentation depends on the rate and volume of bleeding: slow ooze → occult blood loss → iron-deficiency anaemia; brisk bleed → haematemesis (vomiting red blood or coffee-ground material — coffee-ground appearance results from haemoglobin being denatured to haematin by gastric acid) and/or melaena (black, tarry stool — the result of haemoglobin being digested through the entire GI tract and converted to haematin and other pigments by gut bacteria).
These are testable and clinically important — they identify patients who need close monitoring or early surgical/IR referral:
Risk factors for recurrent bleeding [1]:
- Patient already hospitalised (stress, comorbidities, NSAIDs)
- Large ulcer
- Ulcer on posterior D1 (proximity to GDA)
- Ulcer on higher posterior lesser curve (proximity to left gastric artery)
Additional risk factors [2]:
- Age > 60
- Coagulopathy or anticoagulant use
- Hb < 8.0 g/dL on presentation
- Shock on presentation
- Need for blood transfusion
After initial endoscopic haemostasis, continuous vigilance is required:
- Haematemesis or fresh melaena
- Tachycardia (the earliest haemodynamic sign — the body compensates by increasing heart rate before blood pressure drops)
- Falling Hb trend on serial monitoring (anaemia developing despite transfusion)
- Blood in the NG tube [2]
Post-Endoscopic Monitoring
Post-endoscopic monitoring of Hb count is required to identify rebleeding [2]. Check Hb every 6–8 hours for the first 24–48 hours after endoscopic therapy. A drop of > 2 g/dL despite transfusion suggests rebleeding. Combine with clinical signs (tachycardia, hypotension, fresh haematemesis/melaena).
This has been covered in detail in the management section; here is a concise summary of the escalation ladder:
Step 1: Resuscitate → ABC, 2 large-bore IV, fluids, blood if Hb < 7 (or < 9 if high-risk)
Step 2: Endoscopy → Dual therapy (adrenaline 1:10,000 [tamponade + vasoconstriction + platelet aggregation] + heater probe [coaptive effect: pressure + heat] or metal clip) for Forrest Ia, Ib, IIa, IIb [1][3]
- Clean base → start feeding, early discharge [1]
- Injection therapy should NOT be used as monotherapy [2][3]
Step 3: Post-OGD → IV PPI infusion (80 mg stat → 8 mg/h × 72h) — higher pH stabilises clot since platelet function depends on normal pH [3]
Step 4: If rebleed → Re-scope + repeat endoscopic therapy
Step 5: If 2nd endoscopy fails → Surgery or TAE [1][2][3]
Surgery for bleeding ulcer — indications [1]:
- Therapeutic endoscopist not available
- Massive bleeding
- Failed endoscopic therapy
- Rebleed after endoscopic therapy
Surgery = Plication of bleeder + additional procedure [1]:
- DU: V+P (truncal Vagotomy + Pyloroplasty) [1]
- GU: partial gastrectomy [1] (because GU may harbour malignancy)
TAE (transcatheter arterial embolisation): equally effective as surgery for patients who failed endoscopic therapy; fewer complications; should be considered in patients unfit for surgery [2][3]
Limitations of endoscopic haemostasis [3]:
- Massive bleeding obscuring view
- Large bleeding artery ( > 3.2 mm)
- Large ulcer ( > 2 cm)
2. Perforation
When the ulcer erodes through all layers of the bowel wall (mucosa → submucosa → muscularis propria → serosa) and there is no adjacent organ to seal the defect, the luminal contents spill freely into the peritoneal cavity. This causes:
-
Chemical peritonitis (first 4–6 hours) — gastric acid (pH ~1–2), pepsin, bile, and food particles directly irritate the peritoneum. This produces the classic sudden, severe, "thunderclap" epigastric pain and board-like rigidity (reflex tonic contraction of the abdominal wall muscles to splint the inflamed peritoneum).
-
Bacterial peritonitis (after 6 hours) — gut flora colonise the contaminated peritoneal fluid → systemic inflammatory response → sepsis → multi-organ failure if untreated [3].
Most common site: anterior wall of D1 [3] — because the anterior wall faces the free peritoneal cavity with nothing to tamponade or seal the defect (contrast with the posterior wall which is retroperitoneal and abutted by the pancreas).
- Sudden onset severe generalised abdominal pain — the patient can often pinpoint the exact onset time (unlike gradual-onset pain of pancreatitis or cholecystitis) [3]
- Board-like rigidity with patient lying completely still (any movement worsens peritoneal irritation)
- +/- RLQ pain (Valentino's sign): GI contents track down the right paracolic gutter to the RIF, mimicking appendicitis [3]
- Progression: Shock → fever — chemical peritonitis in first 4–6 hours → bacterial peritonitis after 6 hours [3]
- Loss of liver dullness on percussion (Jobert sign) — free gas between the liver and abdominal wall
- Erect CXR: free gas under diaphragm in 60–70% of cases (require sitting upright for ≥ 10 minutes before film) [3]
- CT thorax + abdomen with contrast: rule out other DDx; detect small amounts of free gas and fluid that CXR may miss
- Bloods: CBC (Hb for GIB, WCC), LRFT, T&S, clotting, amylase (↑ due to pancreatic irritation by leaked duodenal contents — but typically a mild elevation; if markedly elevated, suspect acute pancreatitis as the primary diagnosis instead) [3]
- Do NOT attempt OGD: may convert sealed-off perforation to real perforation [3]
Why NOT Do OGD in Suspected Perforation?
Gas insufflation during endoscopy increases intraluminal pressure. If a perforation has been partially sealed by omentum or fibrin, the pressure blows the seal open → converts a contained leak into a free perforation with flooding peritonitis. This is a well-known iatrogenic disaster. If peritonism and pneumoperitoneum are present, go to the operating theatre, not the endoscopy suite [3].
Resuscitation:
- NPO, IV fluids, NG tube decompression
- IV broad-spectrum antibiotics (e.g., Augmentin) — to cover emerging bacterial peritonitis
- IV PPI / H₂ blocker — suppress acid production, limit ongoing chemical peritonitis
Surgery [3]:
| Ulcer Size | Surgical Approach |
|---|---|
| Small DU/GU ( < 2 cm) | Laparoscopic or open (Graham) omental patch repair + peritoneal lavage ( > 6L NS) |
| Large DU | Distal gastrectomy + Billroth II reconstruction +/- controlled duodenostomy |
| Large GU | Wedge excision / partial gastrectomy + Billroth II reconstruction |
- Biopsy ulcer edge for ALL gastric ulcers to rule out malignancy [3]
- H. pylori eradication should be included in the treatment plan [3]
Convert to open if [3]:
- Haemodynamically unstable (shorter operative time)
- Large defect ( > 1 cm) (difficult laparoscopic suture)
- Gross contamination (limited laparoscopic peritoneal lavage)
Post-op management [3]:
- NPO until no signs of leakage and return of bowel function
- Complete full course of antibiotics
- Continue PPI until OGD
- FU OGD 6–8 weeks after surgery for healing + H. pylori status [3]
Prognosis — Boey's Score (3 points) [3]:
| Risk Factor | Points |
|---|---|
| Time from perforation to admission > 24 hours | 1 |
| Pre-op SBP < 100 mmHg | 1 |
| ≥ 1 systemic illness (heart disease, liver disease, renal disease, DM) | 1 |
| Score | Mortality |
|---|---|
| 0 | 0% |
| 1 | 10% |
| 2 | 45.5% |
| 3 | 100% |
3. Gastric Outlet Obstruction (GOO)
GOO from PUD occurs through three mechanisms working in sequence:
-
Acute oedema and spasm — active ulceration at or near the pylorus/D1 causes inflammatory swelling and smooth muscle spasm → functional narrowing. This component is potentially reversible with acid suppression and anti-inflammatory treatment.
-
Chronic fibrosis and scarring — repeated cycles of ulceration and healing deposit collagen in the pyloric/duodenal wall → fixed structural narrowing. This is irreversible and requires mechanical intervention (dilatation or surgery).
-
Gastric atony — prolonged obstruction leads to chronic gastric distension → the gastric wall muscle becomes over-stretched and loses contractile ability → even after the obstruction is relieved, the stomach may not empty properly [2].
- Waxing-and-waning epigastric pain (pain worsens after eating as the obstructed stomach distends, then partially subsides as some content trickles through)
- Repeated non-bilious projectile vomiting of undigested food — "non-bilious" because the obstruction is proximal to the ampulla of Vater (bile enters in D2, but the obstruction is at the pylorus/D1). "Undigested" because the food never reaches the small bowel for digestion. "Projectile" because the stomach builds up high pressure behind the obstruction [3]
- Early satiety, weight loss — the patient cannot eat without vomiting
- Succussion splash — audible splash heard with a stethoscope when shaking the patient's abdomen > 4 hours after the last meal; indicates retained gastric fluid. Pathophysiology: the dilated, atonic stomach retains food and fluid like a water balloon, and shaking it produces the characteristic sloshing [2]
This is a high-yield exam topic — the electrolyte disturbance in GOO is pathognomonic:
Hypochloraemic hypokalaemic metabolic alkalosis +/- paradoxical aciduria [2][3]
Step-by-step mechanism:
- Repeated vomiting of HCl → loss of H⁺ and Cl⁻ → metabolic alkalosis + hypochloraemia
- Volume depletion → kidney activates RAAS → aldosterone → proximal tubule Na⁺ reabsorption in exchange for K⁺ and H⁺ secretion → hypokalaemia + paradoxical aciduria (the kidneys excrete acid despite the body being alkalotic, because Na⁺ salvage takes priority over acid-base balance)
- Cl⁻ deficit prevents the kidney from excreting HCO₃⁻ (because Cl⁻ is needed for HCO₃⁻ exchange in the proximal tubule) → alkalosis is maintained = "chloride-responsive alkalosis"
- Hypokalaemia itself worsens alkalosis: in the distal tubule, K⁺ depletion causes H⁺ to be secreted instead of K⁺, further contributing to paradoxical aciduria and perpetuating alkalosis
- Hypocalcaemia may develop → tetany (alkalosis increases protein-binding of ionised Ca²⁺, reducing the free fraction) [3]
Correction: IV normal saline (0.9% NaCl) + KCl — NS provides both volume and chloride, allowing the kidneys to finally excrete excess bicarbonate. KCl corrects hypokalaemia and enables H⁺/K⁺ exchange to normalise [2].
Why Normal Saline Specifically?
You MUST give NaCl, not dextrose or lactated Ringer's. The root problem is chloride depletion — without adequate chloride delivery to the kidney, the proximal tubule avidly reabsorbs all filtered HCO₃⁻ (because HCO₃⁻ reabsorption is coupled to Cl⁻). Only by replenishing Cl⁻ can the kidney "let go" of HCO₃⁻ and correct the alkalosis. This is why it's called a chloride-responsive metabolic alkalosis.
Step 1: Medical ("Drip and suck")
- NPO, NG tube decompression, IV fluid resuscitation
- Correction of electrolytes — NS + KCl [2]
- IV PPI (e.g., pantoprazole) — reduces acid secretion, may reduce acute inflammatory oedema component [2]
Step 2: OGD after decompression
- Biopsy to exclude malignancy — GOO is malignant until proven otherwise (80% malignant, 20% benign) [3]
Step 3: Definitive treatment
- Benign: Endoscopic balloon dilatation [2] +/- stenting; if fails → surgery
- Malignant: treat underlying CA; palliative bypass (gastrojejunostomy) or SEMS [3]
Surgical options for benign GOO [2]:
- GU: Antrectomy with Billroth I/II reconstruction
- DU: Truncal vagotomy + Antrectomy with Billroth I/II reconstruction
4. Penetration
Penetration is essentially a "sealed perforation" — the ulcer erodes through the full thickness of the bowel wall, but instead of spilling into the free peritoneal cavity, the adjacent organ acts as a plug, preventing free leakage. The ulcer crater extends into the parenchyma of the neighbouring organ.
Order of frequency of penetration [2]: Pancreas > Lesser omentum > Biliary tract > Liver > Greater omentum > Mesocolon > Colon > Vascular structures
Why is the pancreas most common? Because posterior DU sit directly against the head of the pancreas (the pancreatic head is immediately retroperitoneal to D1). As the ulcer bores posteriorly, the first structure it encounters is the pancreas.
- Pain becomes more intense, longer duration
- Shift from typical vague visceral discomfort to localised intense pain radiating to the back — this back radiation occurs because the pancreas is a retroperitoneal organ, and inflammation of retroperitoneal structures causes somatic pain referred to the back (T10–L1 dermatomes)
- Pain is NOT relieved by food or antacids — unlike uncomplicated DU, where food buffers acid and provides temporary relief, in penetration the inflammatory process in the adjacent organ is ongoing and unrelated to intraluminal acid levels [2]
- Secondary complications: pancreatitis (most common — from DU penetrating into pancreas), cholangitis (if penetrating into biliary tract) [2]
- Surgical treatment is NOT recommended [2]
- Conservative management: IV PPI (high-dose acid suppression to halt further ulcer progression), H. pylori eradication if positive, treat the secondary complication (e.g., manage pancreatitis per standard protocol)
- Surgery reserved only for: uncontrollable haemorrhage from vascular erosion, fistula formation that does not resolve, or failure of conservative therapy
5. Fistulisation
Fistulisation represents the most extreme form of penetration — the ulcer erodes completely through the bowel wall AND through the wall of an adjacent hollow organ, creating an abnormal communication (fistula) between two luminal structures.
Types:
- Gastrocolic fistula — stomach → transverse colon (the transverse colon lies immediately inferior to the greater curvature)
- Duodenocolic fistula — duodenum → colon
- Choledochoduodenal fistula — CBD → duodenum (can lead to gallstone ileus via this route)
The symptoms are dramatic because colonic content now refluxes into the stomach:
- Feculent vomiting — the patient vomits material with faecal odour/character (colonic bacteria + faecal matter refluxing into the stomach). This is nearly pathognomonic of a gastrocolic/duodenocolic fistula
- Postprandial diarrhoea — food bypasses the small bowel absorptive surface by directly entering the colon through the fistula → malabsorption + osmotic diarrhoea
- Dyspepsia and weight loss — chronic malabsorption and ongoing inflammation
- Halitosis (faecal breath)
- Nutritional optimisation (patients are often severely malnourished)
- Surgery: resection of the fistulous tract with repair of both organs involved; typically requires partial gastrectomy or duodenal repair + colonic resection
- IV PPI + H. pylori eradication as adjuncts
6.1 Pathophysiology [2]
- Gastric ulcers are associated with pangastritis → atrophy → intestinal metaplasia → dysplasia → adenocarcinoma. This sequence is driven by chronic inflammation and the loss of specialised gastric glands, with compensatory proliferation of metaplastic epithelium that accumulates genetic mutations over time [2].
- Duodenal ulcers are associated with antral-predominant gastritis that spares the acid-secreting body → no atrophy → no metaplasia → does NOT predispose to gastric cancer [2].
- A GU that fails to heal after 12 weeks of adequate therapy may harbour an underlying malignancy that was missed on initial biopsy (sampling error) — this is why non-healing GU > 3 months requires surgery even if initial biopsy is benign [2][3].
DU Does NOT Cause Cancer
This is a crucial distinction. The acid-hypersecretory, antral-predominant gastritis of DU does not produce the atrophy-metaplasia-dysplasia sequence. Only GU (via pangastritis, atrophy, and intestinal metaplasia) carries a cancer risk. This is why all GU must be biopsied and followed to healing, but uncomplicated DU do not need follow-up endoscopy [2].
7. Post-Surgical (Gastrectomy) Complications
When PUD is treated surgically (now rare, but still tested), the altered anatomy creates its own set of problems. These are collectively called "post-gastrectomy syndromes" and are important exam material [3].
| Complication | Mechanism | Clinical Features | Management |
|---|---|---|---|
| Anastomotic leak / duodenal stump blowout | Technical failure at the surgical anastomosis or duodenal stump closure → contents leak into peritoneum | Fever, peritonism, sepsis in the early post-op period | Urgent re-operation; controlled fistula drainage |
| Afferent loop syndrome (Billroth II) | Obstruction or kinking of the afferent (duodenal) limb → bile and pancreatic juice accumulate under pressure → sudden decompression | Severe epigastric pain → sudden bilious vomiting that relieves the pain (the "pressure-release" pattern); abdominal distension [3] | Revise to Roux-en-Y or Braun's enteroenterostomy [3] |
| Efferent loop obstruction (Billroth II) | Kinking, adhesion, or herniation of the efferent limb | Gastric outlet obstruction picture (vomiting, distension) | Surgical revision |
| Marginal (stomal/anastomotic) ulcer | Ulcer at the surgical anastomotic site — caused by acid exposure at the neo-junction, retained gastric antrum (continued gastrin production), incomplete vagotomy, or H. pylori recurrence | Recurrent epigastric pain resembling PUD | Investigate: check for H. pylori, retained antrum, ZES; PPI therapy; revise surgery if refractory |
| Internal hernia (Petersen's defect) | Bowel herniates through the mesenteric defect created during reconstruction → risk of strangulation | Small bowel obstruction; acute abdominal pain | Urgent surgical reduction |
| Complication | Mechanism | Clinical Features | Management |
|---|---|---|---|
| Early dumping syndrome | Loss of pylorus → rapid gastric emptying of hyperosmolar carbohydrates into small bowel → osmotic fluid shift into intestinal lumen → hypovolaemia + distension + release of vasoactive gut hormones (serotonin, VIP, neurotensin) [3] | 15–30 min post-meal: abdominal pain, nausea, vomiting, diarrhoea, vasomotor symptoms (sweating, flushing, dizziness, tachycardia, palpitations) [3] | Small frequent meals, avoid simple carbohydrates; octreotide for refractory cases [3] |
| Late dumping syndrome | Rapid glucose absorption → exaggerated insulin release → reactive (post-prandial hyperinsulinaemic) hypoglycaemia [3] | 2–3 hours post-meal: tremor, diaphoresis, confusion, weakness, hunger [3] | Same dietary advice; glucose for acute episodes |
| Alkaline reflux gastritis | Loss of pyloric sphincter → bile refluxes into gastric remnant → bile acids disrupt the mucosal barrier → chronic gastritis | Burning epigastric pain, bilious vomiting; pain is NOT relieved by antacids (because the problem is bile, not acid) | Prokinetics; revision to Roux-en-Y (diverts bile away from gastric remnant) |
| Gastric stasis / gastroparesis | Vagal denervation (post-vagotomy) → impaired gastric motility | Early satiety, nausea, vomiting, bloating | Prokinetics (metoclopramide, erythromycin) |
| Roux stasis syndrome | Ectopic pacemakers in the Roux limb → net retrograde propulsive activity towards the stomach [3] | Bloating, nausea, vomiting, heartburn, delayed regurgitation | Prokinetics; surgical revision if severe |
| Deficiency | Mechanism | Consequences | Management |
|---|---|---|---|
| Vitamin B12 deficiency | Loss of parietal cells → ↓ intrinsic factor (intrinsic factor is produced by parietal cells and is required for B12 absorption in the terminal ileum); also ↓ gastric acid → impaired B12 release from food proteins [3] | Megaloblastic anaemia, subacute combined degeneration of the spinal cord, peripheral neuropathy | IM B12 every 3 months [3] — oral supplementation is ineffective because the problem is lack of intrinsic factor |
| Iron deficiency | ↓ Gastric acid → impaired conversion of Fe³⁺ (dietary ferric iron) to Fe²⁺ (ferrous iron, the absorbable form); also, bypassing the duodenum (the main site of iron absorption in Billroth II) + chronic blood loss from anastomotic ulcers [3] | Iron-deficiency anaemia (microcytic, hypochromic) | Oral or IV iron supplementation |
| Calcium and Vitamin D deficiency | ↓ Gastric acid → impaired calcium solubilisation; loss of duodenal continuity (Billroth II/Roux-en-Y) → bypass of the main Ca²⁺ and vitamin D absorption site; fat malabsorption → vitamin D (fat-soluble) malabsorption [3] | Osteomalacia, osteopenia, osteoporosis, secondary hyperparathyroidism | Calcium + Vitamin D supplements; DEXA screening |
| Fat-soluble vitamins (A, D, E, K) | Loss of duodenal continuity → reduced mixing of bile/pancreatic enzymes with chyme → fat malabsorption → steatorrhoea → fat-soluble vitamin loss [3] | Night blindness (A), bone disease (D), neuropathy (E), coagulopathy (K) | Vitamin supplementation |
| Steatorrhoea | Poor mixing of bile and pancreatic enzymes with food due to Billroth II/Roux-en-Y anatomy → incomplete fat digestion [3] | Fatty diarrhoea, weight loss, malnutrition | Pancreatic enzyme replacement; dietary fat restriction |
Why IM B12 and Not Oral?
After gastrectomy, the patient has lost the parietal cells that produce intrinsic factor. Without intrinsic factor, oral B12 cannot be absorbed in the terminal ileum (B12-IF complex binds to cubam receptors there). Therefore, B12 must be given by the intramuscular (IM) route, bypassing the GI tract entirely. This is a lifelong requirement [3].
| Complication | Key Pathophysiology | Classic Presentation | Critical Management Point |
|---|---|---|---|
| Haemorrhage | Posterior DU → GDA erosion; GU → left gastric a. | Haematemesis, melaena, shock | Endoscopic dual therapy → IV PPI; surgery if fails |
| Perforation | Anterior DU → free peritoneal spillage | Thunderclap pain, board-like rigidity, pneumoperitoneum | Omental patch + peritoneal lavage; biopsy GU edges |
| GOO | Pyloric fibrosis/oedema → mechanical obstruction | Non-bilious projectile vomiting, succussion splash, hypoCl hypoK metabolic alkalosis | Drip-and-suck → OGD + biopsy (malignant until proven otherwise) |
| Penetration | Posterior DU → pancreas (sealed) | Intense back pain, NOT relieved by food | Conservative — surgery NOT recommended |
| Fistulisation | Through-and-through into adjacent hollow organ | Feculent vomiting (gastrocolic fistula) | Surgery: resection + repair |
| Malignancy | GU → atrophy → metaplasia → dysplasia → CA | Non-healing GU; weight loss | Always biopsy GU; surgery if non-healing > 12 weeks |
High Yield Summary
- Haemorrhage is the leading cause of death from peptic ulcer [3]. Posterior DU erodes into GDA; GU on lesser curvature into left gastric artery.
- Risk factors for recurrent bleeding: patient already hospitalised, large ulcer, posterior D1, higher posterior lesser curve [1].
- Signs of rebleeding: haematemesis, fresh melaena, tachycardia, falling Hb, blood in NG tube [2].
- Perforation: MC anterior wall of D1. Chemical peritonitis (first 4–6h) → bacterial peritonitis (after 6h). Erect CXR shows free gas in 60–70%. Do NOT do OGD [3].
- Boey's Score: delay > 24h (1), SBP < 100 (1), systemic illness (1). Score 3 = 100% mortality [3].
- GOO: hypochloraemic hypokalaemic metabolic alkalosis + paradoxical aciduria. Treat with NS + KCl. GOO is malignant until proven otherwise (80%) [2][3].
- Penetration: most commonly into pancreas. Pain shifts to localised, intense back pain not relieved by food. Surgery NOT recommended [2].
- Fistulisation: gastrocolic fistula → feculent vomiting (pathognomonic).
- Malignant transformation: GU only (via Correa cascade: atrophy → metaplasia → dysplasia → CA). DU does NOT predispose to gastric cancer [2].
- Post-gastrectomy syndromes: early dumping (osmotic shift, 15–30 min), late dumping (reactive hypoglycaemia, 2–3h), afferent loop syndrome (bilious vomiting relieving pain), B12/iron/Ca deficiency.
- IM B12 every 3 months is lifelong after gastrectomy — oral B12 is ineffective without intrinsic factor [3].
Active Recall - Complications of PUD
References
[1] Lecture slides: GC 198. Profuse vomiting of fresh blood and in shock severe upper GI bleeding.pdf (p24 — bleeding peptic ulcer: clean base, therapeutic endoscopy, PPI; p27 — risk factors for recurrent bleeding; p28 — surgery for bleeding ulcer: indications; p29 — choice of additional procedure) [2] Senior notes: felixlai.md (PUD complications and management pp. 395–398 — haemorrhage, perforation, GOO, penetration, fistulisation, prevention; PUD overview p. 386 — relationship with gastric cancer) [3] Senior notes: maxim.md (PUD complications pp. 127–131 — haemorrhage, perforation, GOO, Boey's score, post-op management; Post-gastrectomy syndromes pp. 144–146 — afferent loop, dumping, nutritional deficiencies; UGIB endoscopic therapy p. 53 — dual therapy, PPI infusion)
High Yield Summary
- PUD = mucosal defect through the muscularis mucosae — "no acid, no ulcer" but the balance of aggression vs defence is the true paradigm.
- DU (~75%) > GU (~20%); DU almost always in D1; GU on lesser curvature/antrum.
- Two dominant causes: H. pylori (90–95% DU, 60–80% GU) and NSAIDs — these are synergistic.
- NSAIDs cause ulcers systemically by inhibiting COX-1 → ↓ prostaglandins → ↓ mucus, bicarbonate, blood flow.
- Modified Johnson classification (Types I–V) classifies GU by location and acid status → guides surgical approach.
- "Posterior bleeds, Anterior perforates" — posterior DU erodes into GDA; anterior DU perforates freely.
- DU pain: hunger pain, relieved by food, nocturnal. GU pain: worse with food.
- Up to 70% of NSAID ulcers are silent — first presentation may be haemorrhage or perforation.
- Always biopsy GU to exclude malignancy; DU biopsy is not routine.
- GOO → non-bilious projectile vomiting, succussion splash, hypochloraemic hypokalaemic metabolic alkalosis.
- ZES: suspect if recurrent/refractory ulcers, unusual location (D2/jejunum), no H. pylori/NSAIDs.
- PUD is the most common cause of UGIB [1].
High Yield Summary
- PUD is the most common cause of UGIB — but always consider varices (liver disease), Mallory-Weiss (Hx of forceful vomiting), malignancy (constitutional symptoms), and aorto-enteric fistula (Hx of aortic graft).
- Functional dyspepsia accounts for ~60% of dyspepsia — diagnosis of exclusion after OGD is normal.
- Always do an ECG in acute epigastric pain to exclude inferior MI — the single most dangerous mimic.
- Gastric ulcers must always be biopsied to exclude malignancy — a malignant gastric ulcer looks identical to a benign one.
- GOO is malignant until proven otherwise (80% malignant, 20% benign).
- Valentino's sign: PPU fluid tracking to RIF mimicking appendicitis.
- Aorto-enteric fistula: any patient with Hx of aortic graft + UGIB → CT aortogram urgently.
- "Test and treat" strategy for young patients without alarm features — non-invasive H. pylori testing before endoscopy.
- UGIB differentials by site: Oesophagus (varices, oesophagitis, Mallory-Weiss, CA), Stomach (PUD, gastritis, Dieulafoy, portal hypertensive gastropathy, varices, CA), Duodenum (DU, duodenitis, haemobilia, CA).
High Yield Summary
- OGD is the gold standard for diagnosing PUD — both diagnostic and therapeutic [2][9].
- Alarm features → urgent OGD. No alarm features and age < 55 → "test and treat" with non-invasive H. pylori testing [2].
- ALL patients with endoscopy-diagnosed PUD must be tested for H. pylori — CLO test (1 biopsy) + histology (2 biopsies) from the antrum (+ body to increase sensitivity) [2].
- Stop PPIs ≥ 2 weeks and antibiotics ≥ 4 weeks before H. pylori testing to avoid false negatives.
- UBT is the best non-invasive test for both diagnosis and confirmation of eradication [2].
- Serology cannot distinguish active from past infection — do NOT use it to confirm eradication.
- Forrest classification guides endoscopic therapy: Class I and IIa/IIb → endoscopic dual therapy + IV PPI infusion. Class IIc and III → PPI alone, early discharge [1][2].
- Dual endoscopic therapy: adrenaline injection + heater probe/clip. Adrenaline alone is insufficient [1][3].
- Post-OGD IV PPI infusion (80 mg bolus → 8 mg/h × 72h) — stabilises clot by maintaining pH > 6. For ulcer bleeding ONLY, not varices [3].
- All gastric ulcers must be biopsied (multiple from rim). Follow-up OGD mandatory until healing confirmed. Non-healing GU > 12 weeks → surgery even if biopsy benign [2][3].
- Erect CXR for suspected PPU — sensitivity ~75–80%; if negative but clinical suspicion persists → CT [2][5].
- Elevated urea:creatinine ratio > 100:1 is a clue to UGIB [5].
- Glasgow-Blatchford Score: pre-endoscopy; GBS = 0 → safe outpatient management. Rockall Score: post-endoscopy; predicts mortality [5].
- Avoid endoscopy in suspected perforation — gas insufflation can convert a sealed-off perforation to free perforation [5].
High Yield Summary
- Uncomplicated PUD management = remove the cause + heal the ulcer + prevent recurrence.
- H. pylori eradication is the most important treatment for H. pylori-positive PUD. Standard triple therapy (PPI + amoxicillin + clarithromycin × 14 days) or bismuth quadruple therapy. Confirm eradication with UBT ≥ 4 weeks post-Rx.
- H. pylori ulcers do NOT need maintenance PPI after successful eradication [2]. NSAID and idiopathic ulcers DO.
- Aspirin in bleeding PUD: resume with PPI cover once haemostasis secured — do NOT permanently stop [2][3].
- Ulcer bleed stops spontaneously in ~70–80% [1]. Identify shock and ongoing bleeding.
- Endoscopic dual therapy (adrenaline + heater probe/clip) for Forrest Ia, Ib, IIa, IIb. Injection therapy alone is NOT sufficient [2][3].
- Post-OGD IV PPI infusion (80 mg stat → 8 mg/h × 72h) for high-risk ulcer bleeding. NOT for varices [3].
- Clean base (Forrest III): start feeding, early discharge [1].
- Surgery for bleeding ulcer: plication of bleeder + additional procedure. DU: V+P; GU: partial gastrectomy [1].
- TAE is an alternative to surgery if patient is unfit; equally effective with fewer complications [2].
- Perforation: Small — omental patch; Large DU — gastrectomy + Billroth II; Large GU — wedge excision/gastrectomy. Always biopsy GU edges [3].
- GOO: Drip and suck → OGD + biopsy → malignant until proven otherwise [3].
- Penetration: surgery NOT recommended; treat conservatively [2].
- Elective surgery indications: complicated PUD, refractory to medical Rx (exclude ZES first), non-healing GU > 12 weeks [2][3].
- Boey's Score for PPU prognosis: delay > 24h, SBP < 100, systemic illness (0–3 points) [3].
High Yield Summary
- Haemorrhage is the leading cause of death from peptic ulcer [3]. Posterior DU erodes into GDA; GU on lesser curvature into left gastric artery.
- Risk factors for recurrent bleeding: patient already hospitalised, large ulcer, posterior D1, higher posterior lesser curve [1].
- Signs of rebleeding: haematemesis, fresh melaena, tachycardia, falling Hb, blood in NG tube [2].
- Perforation: MC anterior wall of D1. Chemical peritonitis (first 4–6h) → bacterial peritonitis (after 6h). Erect CXR shows free gas in 60–70%. Do NOT do OGD [3].
- Boey's Score: delay > 24h (1), SBP < 100 (1), systemic illness (1). Score 3 = 100% mortality [3].
- GOO: hypochloraemic hypokalaemic metabolic alkalosis + paradoxical aciduria. Treat with NS + KCl. GOO is malignant until proven otherwise (80%) [2][3].
- Penetration: most commonly into pancreas. Pain shifts to localised, intense back pain not relieved by food. Surgery NOT recommended [2].
- Fistulisation: gastrocolic fistula → feculent vomiting (pathognomonic).
- Malignant transformation: GU only (via Correa cascade: atrophy → metaplasia → dysplasia → CA). DU does NOT predispose to gastric cancer [2].
- Post-gastrectomy syndromes: early dumping (osmotic shift, 15–30 min), late dumping (reactive hypoglycaemia, 2–3h), afferent loop syndrome (bilious vomiting relieving pain), B12/iron/Ca deficiency.
- IM B12 every 3 months is lifelong after gastrectomy — oral B12 is ineffective without intrinsic factor [3].
Hiatus Hernia
A hiatus hernia is the protrusion of a portion of the stomach through the esophageal hiatus of the diaphragm into the thoracic cavity.
Upper Gi Bleed
Upper gastrointestinal bleeding is hemorrhage originating from a source proximal to the ligament of Treitz, commonly caused by peptic ulcers, esophageal varices, or Mallory-Weiss tears.