Turner Syndrome (45,x)
Turner syndrome is a chromosomal disorder affecting females who have a complete or partial absence of one X chromosome (45,X), typically presenting in childhood or adolescence with short stature, delayed puberty, ovarian dysgenesis, and characteristic features such as webbed neck and broad chest.
Turner Syndrome (45,X) — Paediatric Notes
Turner syndrome (TS) is a chromosomal disorder affecting phenotypic females characterised by the complete or partial absence of one X chromosome. The classic karyotype is 45,X (monosomy X), but numerous mosaic and structural X-chromosome variants exist.
Breaking down the name: "Turner" is eponymous (after Henry Turner, who described it in 1938). The karyotype notation "45,X" means 45 total chromosomes with only a single X sex chromosome — instead of the usual 46,XX.
The unifying concept is haploinsufficiency of genes on the X chromosome — having only one working copy of genes that normally require dosage from two X chromosomes for normal development [1].
High Yield – GC Lecture Point
"Clinical features of Turner syndrome — Haploinsufficiency of genes on X chromosome" [1]. This is the core pathophysiological principle: many X-linked genes escape X-inactivation and normally require expression from both X chromosomes. Loss of one copy → haploinsufficiency → the phenotypic features of TS.
| Feature | Detail |
|---|---|
| Incidence | ~1 in 2,500 live-born females [2] |
| Conception frequency | Much higher — > 95% of 45,X conceptuses result in early miscarriage [2] |
| Contribution to miscarriage | Accounts for 7–10% of all spontaneous abortions [2] |
| Survival to term | Only ~1% of 45,X embryos survive to term [2] |
| Racial/ethnic variation | Occurs across all ethnicities with similar frequency |
| De novo vs inherited | Majority are de novo [2]; low recurrence risk for subsequent pregnancies [2] |
| Paternal origin | Paternal non-disjunction accounts for ~70% of cases (i.e., the lost X is usually the paternal one) [3] |
Why does 45,X cause so much early pregnancy loss? The X chromosome carries genes critical for placental function, cardiac development, and lymphatic development. Haploinsufficiency of these genes causes severe hydrops fetalis and cystic hygroma in utero → the vast majority of affected embryos cannot survive the first trimester.
Increased detection by antenatal ultrasound (e.g., oedema, cystic hygroma, increased nuchal translucency) means many cases are now identified prenatally [2].
3. Anatomy and Function — Relevant Developmental Biology
- In a normal 46,XX female, one X chromosome is randomly inactivated in each cell (Lyon hypothesis) to equalise X-linked gene dosage between males (46,XY) and females (46,XX).
- However, not all genes on the X chromosome are inactivated. Approximately 15–25% of genes on the X chromosome escape X-inactivation and are normally expressed from both X chromosomes.
- In Turner syndrome, these "escape genes" have only one functional copy → haploinsufficiency → phenotypic consequences.
The reason for short stature in Turner syndrome is due to abnormalities of the SHOX gene (Short Stature Homeobox-containing gene) on the X chromosome [2].
- SHOX is located in the pseudoautosomal region 1 (PAR1) at the tip of the short arm of the X chromosome (Xp22.33).
- PAR1 genes escape X-inactivation — both copies are normally active.
- SHOX is a transcription factor critical for long bone growth — it regulates chondrocyte proliferation and differentiation in growth plates.
- In 45,X: only one copy of SHOX → haploinsufficiency → impaired linear growth → short stature.
- This occurs vice versa in Klinefelter syndrome (47,XXY), where additional copies of SHOX are responsible for tall stature [2].
- The second X chromosome is required for maintenance of oocytes and ovarian follicles (not for initial gonadal differentiation — that is determined by SRY on the Y chromosome, which is absent).
- In 45,X: gonads initially develop as ovaries with normal primordial follicles in early fetal life, but accelerated atresia of oocytes occurs from mid-gestation onwards → by birth or early childhood the ovaries are replaced by fibrous "streak gonads" (bands of connective tissue without functional follicles).
- This is termed ovarian dysgenesis → the fundamental endocrine consequence is premature ovarian insufficiency (POI).
- Genes on the X chromosome are involved in lymphatic vessel development.
- Haploinsufficiency → lymphatic hypoplasia/dysplasia → lymphatic obstruction in utero → fetal hydrops, cystic hygroma, peripheral lymphoedema.
- Postnatally, residual lymphatic dysfunction manifests as peripheral lymphoedema of hands and feet (especially in neonates) and webbed neck (from resolved cystic hygroma).
- Left-sided cardiac structures are particularly affected (mechanism not fully understood but possibly related to abnormal neural crest cell migration and haemodynamic consequences of lymphatic dysfunction in utero).
- Key associations: bicuspid aortic valve, coarctation of the aorta, aortic root dilatation.
4. Aetiology and Karyotypic Classification
GC Exam High Yield
Know the major karyotypic subtypes, their approximate frequencies, and clinical implications — especially the risk of gonadoblastoma with Y-chromosome material.
| Karyotype | Frequency | Key Points |
|---|---|---|
| 45,X (classic monosomy X) | ~40–50% [3] | Most severe phenotype; complete absence of one X chromosome |
| 45,X mosaicism | ~30% [3] | Two or more cell lines with different genotypes from a single fertilised egg |
| — 45,X/46,XX | 15–25% [3] | Milder phenotype; may have spontaneous puberty |
| — 45,X/47,XXX or 45,X/46,XX/47,XXX | ~3% [3] | |
| — 45,X/46,XY (mixed gonadal dysgenesis) | 10–12% [3] | Risk of gonadoblastoma |
| Structural abnormalities of X | ~15% [3] | |
| — Isochromosome Xq [46,X,i(Xq)] | ~10% [3] | Two copies of long arm, no short arm → loss of SHOX |
| — Xp deletion [46,X,del(Xp)] | Variable | Loss of short arm → SHOX haploinsufficiency |
| — Ring X [46,X,r(X)] | Rare | May have more severe phenotype if XIST not present |
| — Marker chromosome | Rare | Must check for Y-material |
A) Classic 45,X (~50%)
- Majority de novo [2]
- Caused by loss of entire or portion of X chromosome that includes the tip of the short arm [2] (where SHOX and other escape genes reside)
- Mechanism: non-disjunction during parental meiosis (usually paternal → the lost chromosome is the paternal X or Y) or anaphase lag (chromosome lost during early mitotic division of the zygote)
B) Mosaicism (~30%)
- Mosaicism refers to the presence of two or more populations of cells with different genotypes in an individual who has developed from a single fertilised egg [3]
- Arises from post-zygotic mitotic errors (non-disjunction or anaphase lag after fertilisation)
- Clinical significance: the proportion and tissue distribution of 45,X cells determines phenotypic severity → mosaic patients often have milder features and may have spontaneous puberty and even fertility
C) Structural X Abnormalities (~15%)
- Isochromosome Xq [46,X,i(Xq)]: The X chromosome replicates the long arm on both sides, losing the short arm entirely → loss of SHOX and other Xp genes
- Xp deletion: Partial deletion of the short arm → SHOX haploinsufficiency + variable other gene loss
- Ring X [46,X,r(X)]: Circularised X; if XIST locus is lost, the ring X fails to inactivate → functional disomy for some genes → can paradoxically cause a more severe/atypical phenotype with intellectual disability
D) Y-Chromosome Material
- 10% of individuals with Turner syndrome phenotype may have mosaicism involving a cell line containing Y chromosome material [3]
- Associated with increased risk of germ cell tumours (gonadoblastoma) [3]
- Clinical implication: All patients with Turner syndrome should be screened for Y-chromosome material (by FISH or PCR). If present → prophylactic gonadectomy is recommended due to ~15–30% lifetime risk of gonadoblastoma in dysgenetic gonads containing Y-material.
Must Know for Exams
Do not forget: if Y-chromosome material is found in a Turner syndrome patient → prophylactic gonadectomy is indicated due to risk of gonadoblastoma. This is a commonly tested point.
Understanding why each feature occurs is critical. The overarching mechanism is haploinsufficiency of X-linked genes that escape X-inactivation.
6. Clinical Features
Most commonly (> 90%) short stature and POI (premature ovarian insufficiency) [2].
The clinical features can present at different ages — from the antenatal period through to adulthood. In paediatrics, we focus on the neonatal, childhood, and adolescent presentations.
6.2 Symptoms (with pathophysiological basis)
| Symptom / Presentation | Pathophysiological Basis |
|---|---|
| Swelling of hands and feet (lymphoedema) | Lymphatic hypoplasia → impaired lymphatic drainage → peripheral oedema; classically dorsum of hands and feet |
| Detected antenatally on ultrasound (increased nuchal translucency, cystic hygroma, hydrops fetalis) | Lymphatic obstruction in utero → fluid accumulation; increased detection by antenatal US (e.g., oedema, cystic hygroma) [2] |
| Symptom | Pathophysiological Basis |
|---|---|
| Short stature (the most consistent feature, > 95%) [2] | SHOX gene haploinsufficiency → impaired chondrocyte proliferation in growth plates → reduced long bone growth [2]. Mean adult height without treatment ~143 cm (20 cm below average). Growth failure becomes evident by age 3–5 years and worsens without pubertal growth spurt |
| Learning difficulties with normal intelligence [2] | Haploinsufficiency of X-linked neurodevelopmental genes → specific pattern: normal verbal IQ, impaired visuospatial processing, mathematics, social cognition, and executive function (not global intellectual disability) |
| Recurrent otitis media / hearing problems | Abnormal skull base anatomy → eustachian tube dysfunction → conductive hearing loss; also risk of sensorineural hearing loss in adolescence/adulthood |
| Feeding difficulties (infancy) | High-arched palate, retrognathia → difficulty with latch/feeding |
| Symptom | Pathophysiological Basis |
|---|---|
| Delayed puberty / absent puberty [2] | Ovarian dysgenesis (95%) → streak gonads with no functional follicles → no oestrogen production → failure to enter puberty spontaneously |
| Primary amenorrhoea | No cyclical oestrogen/progesterone from absent ovarian function → no endometrial development → no menses |
| Infertility | Result in POI; pregnancy possible with IVF using donated ova [2] |
| Poor self-esteem / psychosocial difficulties | Short stature + delayed puberty relative to peers → body image concerns; visuospatial and social cognition difficulties compound this |
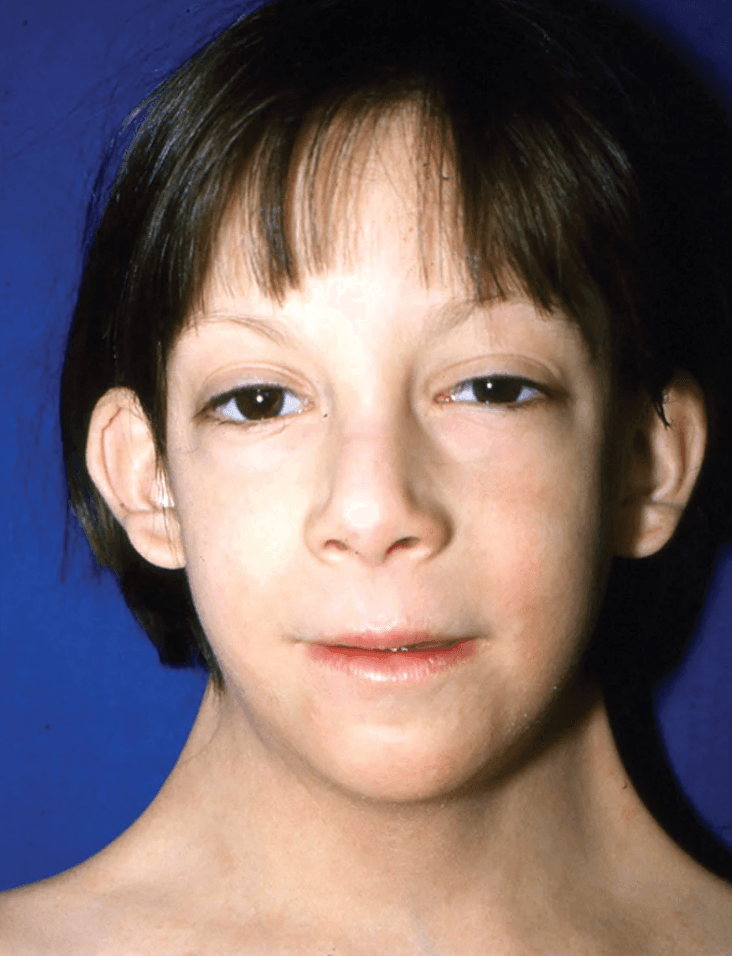
6.3 Signs (with pathophysiological basis)
The signs of Turner syndrome are grouped by system. Each is explained mechanistically.
| Sign | Frequency | Pathophysiological Basis |
|---|---|---|
| Low posterior hairline | 42% [2] | Residual effect of resolved fetal cystic hygroma → skin redundancy at occiput |
| Short webbed neck (pterygium colli) | 25–40% [2] | Resolved cystic hygroma → excess nuchal skin folds draping from mastoid to acromion |
| Low-set ears | Common [2] | Abnormal craniofacial skeletal development (SHOX and other gene effects) |
| Narrow, high-arched palate | 38% [2] | Altered craniofacial growth; contributes to dental crowding and feeding difficulty |
| Retrognathia (micrognathia) | 60% [2] | Underdevelopment of mandible — related to SHOX and craniofacial developmental gene haploinsufficiency |
| Epicanthal folds | Variable | Midface hypoplasia |
| Myopia and strabismus | Variable [2] | Orbital/extraocular muscle developmental effects |
| Sign | Frequency | Pathophysiological Basis |
|---|---|---|
| Short stature | 95% [2] | SHOX haploinsufficiency (see above) |
| Square "shield" chest with widely spaced nipples | Common [2] | Broad thorax with lateral displacement of nipples — skeletal growth pattern from SHOX effects |
| Cubitus valgus (increased carrying angle of elbow) | 47% [2] | SHOX haploinsufficiency affecting skeletal proportions of the upper limb |
| Madelung deformity of wrist | Variable [2] | Abnormal growth of the distal radius (premature fusion of the volar-ulnar physis) → dorsal prominence of distal ulna; directly linked to SHOX haploinsufficiency [2] |
| Shortened 4th metacarpal | Common [2] | Premature epiphyseal fusion in 4th metacarpal — SHOX gene effect |
| Spoon-shaped, hypoplastic nails | 13% [2] | Nail matrix development affected by lymphatic/vascular gene haploinsufficiency |
| Genu valgum (knock-knees) | Variable [2] | SHOX-mediated skeletal disproportion |
| Scoliosis | ~10% | Vertebral growth plate effects |
| Lymphoedema of hands/feet | Neonatal (may persist) | Lymphatic hypoplasia (see above) |
Clinical Pearl
Madelung deformity ("Madelung" = named after Otto Madelung) is a bayonet-shaped deformity of the wrist caused by premature fusion of the volar-ulnar portion of the distal radial growth plate. It is strongly associated with SHOX haploinsufficiency and can occur in Turner syndrome, Léri-Weill dyschondrosteosis, and isolated SHOX mutations.
Congenital heart defects are present in a significant proportion and are the major cause of morbidity and mortality.
| Sign / Anomaly | Frequency | Pathophysiological Basis |
|---|---|---|
| Bicuspid aortic valve (BAV) | 10–15% [2] | Abnormal aortic valve morphogenesis; usually asymptomatic in childhood but may lead to aortic stenosis/regurgitation over time |
| Coarctation of the aorta (CoA) | ~10% [2] | Discrete narrowing of descending aorta near ductus insertion; associated with Turner syndrome [4]; may present with neonatal heart failure and shock if duct-dependent, or with upper limb hypertension + radiofemoral delay if non-duct-dependent |
| Aortic stenosis (AS) | Variable [2] | Secondary to BAV or independent |
| Aortic root dilatation | 8–28% [2] | Connective tissue/vascular wall abnormality → progressive aortic root enlargement → risk of dissection |
| Aortic dissection | 2.5% [2] | Consequence of aortic root dilatation + intrinsic aortic wall abnormality; a major cause of sudden death in TS — risk factors include BAV, CoA, and hypertension |
| Hypertension | ~20% [2] | Multifactorial: may be due to CoA, renal anomalies, or vasculopathy; essential hypertension also more common |
Why is CoA associated with Turner syndrome? The mechanism is not completely understood, but is thought to involve: (1) abnormal neural crest cell migration affecting aortic arch development, (2) haemodynamic consequences of in utero lymphatic obstruction and oedema affecting aortic isthmus flow, and (3) possibly SHOX-related effects on vascular smooth muscle. In Ryan Ho Cardiology notes: "Coarctation of Aorta — Cause: majority sporadic but associated with Turner syndrome" [4].
Signs of CoA to look for on examination:
| Sign / Feature | Frequency | Pathophysiological Basis |
|---|---|---|
| Ovarian dysgenesis → streak gonads | 95% [2] | Accelerated oocyte atresia from haploinsufficiency of X-linked ovarian maintenance genes → no functional follicles |
| Premature ovarian insufficiency (POI) | > 90% | Consequence of ovarian dysgenesis → absent oestrogen → no breast development, no menarche |
| Hypothyroidism | 25–30% [2] | Due to Hashimoto thyroiditis [2] — autoimmune thyroiditis from increased autoimmune predisposition in TS (mechanism: X-linked immune regulatory genes → haploinsufficiency → immune dysregulation) |
| Diabetes mellitus | 2–4× increased risk [2] | Both type 1 (autoimmune) and type 2 (insulin resistance, often related to increased adiposity and metabolic syndrome) |
| Impaired glucose tolerance | Common | Insulin resistance from altered body composition |
Understanding Hypogonadism in Turner Syndrome
Turner syndrome causes primary hypogonadism (gonadal failure) [5]:
- Low oestrogen/testosterone (from non-functional streak gonads)
- With appropriately elevated FSH/LH (the pituitary is working normally and senses the low sex steroids → increases gonadotrophins in an attempt to stimulate the non-functioning gonads)
This is in contrast to secondary hypogonadism (e.g., from pituitary failure), where FSH/LH would be inappropriately low/normal [5].
This distinction is commonly tested. Turner's Syndrome in females (45,XO) → primary hypogonadism → appropriately increased gonadotrophins [5].
| Feature | Frequency | Basis |
|---|---|---|
| Horseshoe kidney | ~10% | Abnormal renal embryogenesis (failure of kidney ascent and rotation) |
| Duplex collecting system | Variable | Ureteric bud duplication |
| Renal malrotation | Variable | |
| Hydronephrosis | Secondary to structural anomalies |
Turner syndrome has a strong predisposition to autoimmune conditions (likely from X-linked immune regulatory gene haploinsufficiency):
- Hashimoto thyroiditis (most common autoimmune association) [2]
- Coeliac disease (~6%)
- Inflammatory bowel disease
- Type 1 diabetes mellitus
- Alopecia areata
| Feature | Basis |
|---|---|
| Multiple pigmented naevi | Increased frequency — mechanism unclear |
| Recurrent otitis media → conductive hearing loss | Eustachian tube dysfunction from craniofacial anatomy |
| Sensorineural hearing loss (progressive) | X-linked inner ear gene effects; becomes significant in young adulthood |
| Osteoporosis | Oestrogen deficiency from POI → inadequate bone mineralisation |
| Metabolic syndrome | Increased visceral adiposity, insulin resistance, dyslipidaemia |
| Age | Key Features |
|---|---|
| Antenatal | Increased nuchal translucency, cystic hygroma, hydrops fetalis, coarctation on fetal echo |
| Neonate | Lymphoedema of hands/feet, webbed neck, low hairline, congenital heart disease |
| Infancy/Early childhood | Short stature (becomes apparent ~age 3–5), feeding difficulties, recurrent otitis media |
| School-age | Growth failure, visuospatial/maths learning difficulties, normal verbal intelligence |
| Adolescence | Delayed/absent puberty, primary amenorrhoea, short stature without pubertal growth spurt |
| Adult | Infertility, cardiovascular complications (aortic dilatation/dissection, HTN), osteoporosis, metabolic syndrome, autoimmune disease |
8. Classification
This is the primary classification — classic 45,X vs mosaic vs structural abnormality.
| Category | Description |
|---|---|
| Classic / severe | Full 45,X karyotype; complete phenotype with short stature, streak gonads, multiple dysmorphic features, cardiac anomalies |
| Mosaic / milder | 45,X/46,XX or other mosaic; may have fewer dysmorphic features; ~15–20% may undergo spontaneous puberty; occasional spontaneous fertility |
| With Y-material | 45,X/46,XY or marker chromosome with Y material; risk of virilisation and gonadoblastoma |
9. Approach to the Child with Suspected Turner Syndrome
- Neonate: lymphoedema, webbed neck, congenital heart disease (especially CoA or BAV)
- Child: unexplained short stature (especially girl with declining growth velocity and no other explanation), recurrent otitis media
- Adolescent: delayed puberty or primary amenorrhoea
- Any age: combination of characteristic dysmorphic features
- Growth history — plot on growth chart; when did parents first notice short stature?
- Developmental milestones — usually normal motor/language; ask about school performance (maths, spatial tasks)
- Pubertal history — any breast development? Menarche?
- Cardiac symptoms — exercise intolerance, chest pain, syncope (think CoA, AS)
- Hearing — recurrent ear infections, hearing difficulties
- Family history — height of parents (to calculate mid-parental height and assess whether short stature is familial or pathological)
- Antenatal history — any abnormalities on antenatal scan?
- Feeding history (infancy) — difficulty latching, GORD
- Psychosocial — self-esteem, peer relationships, school difficulties
- Auxology: height (plot on TS-specific growth chart), weight, BMI, arm span, sitting height
- Dysmorphic features: webbed neck, low hairline, shield chest, cubitus valgus, shortened 4th metacarpal, nail hypoplasia
- Cardiovascular: blood pressure in all four limbs (looking for CoA gradient), murmurs, radiofemoral delay
- Pubertal staging: Tanner staging (breast, pubic hair)
- Skin: pigmented naevi, lymphoedema
- ENT: otoscopy (middle ear effusion)
- Spine: scoliosis
- Abdominal: renal ballotement (horseshoe kidney)
Communication and Family-Centred Care in Paediatrics
When discussing a diagnosis of Turner syndrome:
- Use clear, age-appropriate language with the child and family
- Emphasise that intelligence is usually normal — reassure about school capability (specific support may be needed for maths/spatial tasks)
- Discuss growth hormone treatment and its potential to improve adult height
- Discuss puberty induction at the appropriate time (usually ~age 11–12)
- Address fertility sensitively — discuss options for the future (egg donation, adoption)
- Connect family with Turner syndrome support groups
- Involve a multidisciplinary team (paediatric endocrinologist, cardiologist, audiologist, psychologist, geneticist)
High Yield Summary
Turner Syndrome (45,X) — Key Points for Exams:
- Definition: Phenotypic female with complete or partial loss of one X chromosome; classic karyotype 45,X
- Pathophysiology: Haploinsufficiency of genes on the X chromosome that escape X-inactivation [1]
- Incidence: ~1/2,500 live-born females; > 95% of 45,X conceptions miscarry [2]
- Karyotypes: 45,X (~50%); mosaic (~30%); structural X abnormalities (~15%); 10% may have Y material → risk of gonadoblastoma [3]
- SHOX gene haploinsufficiency → short stature (> 95%) and skeletal anomalies (cubitus valgus, Madelung deformity, shortened 4th metacarpal) [2]
- Ovarian dysgenesis (95%) → streak gonads → POI → delayed/absent puberty, primary amenorrhoea, infertility [2]
- Lymphatic hypoplasia → neonatal lymphoedema, cystic hygroma, webbed neck
- Cardiac: bicuspid AV (10–15%), CoA (~10%), aortic root dilatation (8–28%), dissection (2.5%), HTN (20%) [2][4]
- Autoimmune: Hashimoto thyroiditis (25–30%); coeliac disease; DM (2–4× risk) [2]
- Primary hypogonadism: Low oestrogen + appropriately elevated FSH/LH (↔ secondary hypogonadism = low FSH/LH) [5]
- Learning difficulties with normal intelligence — visuospatial/maths deficits [2]
- Y-material screening: If found → prophylactic gonadectomy (gonadoblastoma risk)
- Low recurrence risk [2]
Active Recall - Turner Syndrome (45,X)
[1] Lecture slides: GC 151. The malformed child hereditary syndromes and anomalies.pdf (Slide: "Clinical features of Turner syndrome — Haploinsufficiency of genes on X chromosome") [2] Senior notes: Adrian Lui Pediatrics Notes.pdf (p506, Turner Syndrome section) [3] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (p847, Turner Syndrome Etiology section) [4] Senior notes: Ryan Ho Cardiology.pdf (p190, Coarctation of Aorta section) [5] Senior notes: Block A - I keep on bumping into people on my side_ pituitary tumours; hypopituitarism.pdf (Hypogonadism section)
Differential Diagnosis of Turner Syndrome (45,X)
Turner syndrome does not typically enter a "differential diagnosis" in the classical sense of a single presenting complaint — because it is a multi-system syndrome that presents differently at different ages. Instead, we approach the DDx from the perspective of the presenting feature(s) that raise suspicion for TS, and then differentiate TS from other conditions that share those features.
The three main clinical "entry points" in paediatrics are:
- Short stature in a girl (most common presentation in childhood)
- Delayed/absent puberty or primary amenorrhoea (most common presentation in adolescence)
- Dysmorphic neonate with lymphoedema, webbed neck, or congenital heart disease
We will cover the DDx systematically for each presentation.
This is the most common way Turner syndrome is picked up in childhood. Short stature affects > 95% of Turner syndrome patients [2] due to SHOX gene haploinsufficiency [2]. However, short stature is extremely common and has many causes.
GC Lecture – High Yield Exam Point
"Karyotype in girls" is listed as a key investigation for short stature workup [6]. The GC growth and development lecture explicitly states: "Female with complete or partial absence of one X chromosome may have features of Turner syndrome including short stature, broad chest, widely spaced nipples, webbed neck, increase in carrying angle, and other system disorders such as cardiac defects, renal anomalies and hypothyroidism" [6]. This means: karyotype is indicated in any girl with unexplained short stature, even in the absence of classic dysmorphic features (because mosaic TS can be phenotypically subtle).
| Category | Condition | Key Distinguishing Features from Turner Syndrome |
|---|---|---|
| Normal variant | Familial short stature (FSS) | Both parents short; child's height appropriate for mid-parental height; normal growth velocity; bone age = chronological age; normal puberty; no dysmorphic features |
| Constitutional delay of growth and puberty (CDGP) | Delayed bone age; family history of "late bloomers"; eventual catch-up growth and normal adult height; delayed but spontaneous puberty; no dysmorphic features | |
| Endocrine | Growth hormone deficiency (GHD) | Proportionate short stature with increased truncal adiposity; delayed bone age; low IGF-1; poor growth velocity; midline defects possible; no webbed neck/shield chest; confirmed by provocative GH testing |
| Hypothyroidism | Lethargy, constipation, cold intolerance, delayed reflexes, goitre (in acquired); markedly delayed bone age; low T4/high TSH; Note: hypothyroidism itself is a feature of TS (25–30% due to Hashimoto) [2] — so it can coexist | |
| Cushing syndrome | Weight gain, growth arrest, striae, buffalo hump, moon face, hypertension; ↑24h urinary cortisol | |
| Precocious puberty (if untreated) | Initially tall then premature epiphyseal fusion → short adult height; advanced bone age; early secondary sexual characteristics — opposite to TS where puberty is absent/delayed | |
| Chromosomal / Syndromic | Noonan syndrome | "Turner-like features" [7] — short stature, ptosis, downslanting palpebral fissures, low-set ears, hypertelorism, webbing of neck, low hairline, shield-like chest, cubitus valgus, cryptorchidism (in males) [7]. KEY distinction: Noonan affects both sexes (autosomal dominant, RAS-MAPK pathway mutations); right-sided cardiac lesions (valvular PS with thick dysplastic valve cusps, ASD, hypertrophic cardiomyopathy) [7] vs Turner's left-sided cardiac lesions (CoA, BAV, AS) [7]; normal karyotype in Noonan |
| Down syndrome (trisomy 21) | Short stature + hypotonia + characteristic facial features (epicanthal folds, upslanting palpebral fissures, flat nasal bridge, protruding tongue); AVSD/VSD; intellectual disability; confirmed by karyotype 47,XX,+21 or 47,XY,+21 | |
| SHOX haploinsufficiency (isolated) | Accounts for 2.2–4.2% of idiopathic short stature (ISS) [2]; may not have any characteristic signs (poor genotype-phenotype correlation) [2]; Leri-Weill dyschondrosteosis (LWD) = SHOX deletion + Madelung deformity; Langer mesomelic dysplasia (LMD) = homozygous SHOX loss → severe short stature; inheritance similar to autosomal disorders (located in pseudoautosomal region, PAR) [2] | |
| Skeletal dysplasia | Achondroplasia | Disproportionate short stature (rhizomelic shortening of limbs); macrocephaly with frontal bossing; trident hand; normal intelligence; FGFR3 mutation |
| Hypochondroplasia | Milder disproportionate short stature; less obvious than achondroplasia; FGFR3 mutation | |
| Chronic disease | Coeliac disease, IBD, CKD, CF, poorly controlled asthma on steroids | Associated with poor nutrition, chronic inflammation, or medication effect → growth failure; screen with relevant investigations; coeliac disease itself is more common in TS (~6%) |
| Nutritional | Malnutrition / psychosocial deprivation | Low weight-for-height; catch-up growth when nutrition/environment improves; history of food insecurity or neglect |
| Iatrogenic | Chronic corticosteroid use | Growth suppression from GC effect on growth plate; Cushingoid features; drug history is key |
Critical Exam Distinction: Noonan vs Turner
This is one of the most commonly tested comparisons in paediatric exams. Both have webbed neck, short stature, shield chest, and cubitus valgus.
| Feature | Turner Syndrome | Noonan Syndrome |
|---|---|---|
| Sex | Phenotypic females only | Both sexes |
| Karyotype | Abnormal (45,X or variant) | Normal karyotype |
| Genetics | X-chromosome monosomy/structural defect | AD; RAS-MAPK pathway (PTPN11 most common) |
| Cardiac lesions | Left-sided: CoA, BAV, AS [7] | Right-sided: Pulmonary valve stenosis (dysplastic), ASD, HCM [7] |
| Gonads | Streak gonads / ovarian dysgenesis | Usually normal ovaries in females; cryptorchidism in males |
| Intelligence | Normal intelligence, learning difficulties [2] | Mild-moderate intellectual disability in ~25% |
| Lymphoedema | Prominent (neonatal) | Can occur but less characteristic |
II. Differential Diagnosis When Presenting with Delayed Puberty / Primary Amenorrhoea
Ovarian dysgenesis (95%) resulting in premature ovarian insufficiency (POI) [2] is the hallmark. But delayed puberty and primary amenorrhoea have a broad differential.
Definitions (paediatric context):
- Delayed puberty: No breast development (Tanner B2) by age 13 in girls; no testicular enlargement by age 14 in boys
- Primary amenorrhoea: No menarche by age 15 (with secondary sexual characteristics present) OR by age 13 (without any secondary sexual development)
The DDx is best organised by the HPG (hypothalamic-pituitary-gonadal) axis level:
The gonad itself has failed. The pituitary appropriately increases gonadotrophins.
Turner's Syndrome in females (45,XO) → primary hypogonadism → appropriately increased gonadotrophins [5].
| Condition | Key Distinguishing Features |
|---|---|
| Turner syndrome (45,X) | Characteristic dysmorphic features; short stature; gonadal dysgenesis → POI [2]; abnormal karyotype |
| 46,XX gonadal dysgenesis (Swyer-like in 46,XX) | Phenotypic female; normal height (no SHOX haploinsufficiency because both X present but gonadal gene mutations, e.g., BMP15, FOXL2); streak gonads; primary amenorrhoea; normal karyotype 46,XX |
| 46,XY complete gonadal dysgenesis (Swyer syndrome) | Phenotypic female (because no functioning testes → no AMH → Müllerian structures develop; no testosterone → female external genitalia); tall stature; streak gonads; risk of gonadoblastoma; karyotype 46,XY |
| Autoimmune oophoritis | May have other autoimmune conditions (Addison disease, hypothyroidism); anti-ovarian antibodies; no dysmorphic features |
| Chemotherapy / radiation-induced gonadal failure | History of cancer treatment; iatrogenic |
| Galactosaemia | Neonatal presentation with jaundice, hepatomegaly, cataracts, E. coli sepsis; long-term ovarian failure; galactose-1-phosphate uridylyltransferase deficiency |
Gonadal dysgenesis (43%) is the most common cause of primary amenorrhoea — the majority being Turner syndrome and Swyer syndrome [8].
The pituitary or hypothalamus has failed. Gonadotrophins are inappropriately low or normal [5].
| Condition | Key Distinguishing Features |
|---|---|
| Constitutional delay of growth and puberty (CDGP) | 14% of primary amenorrhoea [8]; family history of late puberty; delayed bone age; diagnosis of exclusion; eventual spontaneous puberty |
| Kallmann syndrome | GnRH deficiency + anosmia/hyposmia (failure of GnRH neurons to migrate from olfactory placode); may have midline defects (cleft lip/palate); X-linked, AD, or AR |
| Functional hypothalamic amenorrhoea | Almost all hypothalamic causes are functional [8]; associated with eating disorder, excessive exercise, and stress [8]; low body weight; reversible with weight gain |
| Hyperprolactinaemia | Galactorrhoea, persistent headache, bitemporal hemianopia [8] (if macroadenoma); ↑prolactin suppresses GnRH → ↓FSH/LH |
| Hypopituitarism | Due to sellar masses, neurosurgery, infection [8]; may involve multiple pituitary hormone deficiencies |
| Thalassaemia major with iron overload | Secondary amenorrhoea due to hypogonadotrophic hypogonadism, due to haemosiderosis of the pituitary [5]; history of chronic transfusion |
Primary vs Secondary Hypogonadism – Summary
| Primary (Gonadal failure) | Secondary (Hypothalamic/Pituitary) | |
|---|---|---|
| FSH/LH | Appropriately HIGH [5] | Inappropriately LOW/normal [5] |
| Oestrogen | Low | Low |
| Examples | Turner syndrome, Klinefelter syndrome [5] | Kallmann, hyperprolactinaemia, iron overload |
This axis-level localisation is the first step in working up delayed puberty/amenorrhoea. A single FSH level can immediately categorise the problem.
These patients may have normal ovarian function and normal secondary sexual characteristics but fail to menstruate due to a structural problem.
| Condition | Key Distinguishing Features |
|---|---|
| Müllerian agenesis (MRKH syndrome) | 15% of primary amenorrhoea [8]; normal 2° sexual characteristics; no cervix/vagina/uterus (or rudimentary); normal ovarian function; testosterone in female range + pubic/axillary hair (distinguishes from androgen insensitivity); 46,XX karyotype |
| Complete androgen insensitivity syndrome (CAIS) | 46,XY phenotypic female; normal breast development (aromatisation of testosterone → oestrogen); absent/sparse pubic/axillary hair (androgen-dependent); blind vaginal pouch; absent uterus (AMH from testes causes Müllerian regression); inguinal testes on examination |
| Transverse vaginal septum | Associated with cyclical pelvic pain due to cryptomenorrhoea [8]; haematocolpos on imaging; normal secondary sexual characteristics |
| Imperforate hymen | Cyclical pelvic pain; bulging bluish membrane at introitus; haematocolpos |
A neonate with webbed neck, lymphoedema, or congenital heart disease may have:
| Condition | Key Distinguishing Features |
|---|---|
| Turner syndrome | Female; lymphoedema of hands/feet; webbed neck; left-sided cardiac lesions (BAV, CoA) [7]; karyotype 45,X or variant |
| Noonan syndrome | Both sexes; "Turner-like features" [7]; right-sided cardiac lesions (PS, HCM) [7]; normal karyotype; AD inheritance (RAS-MAPK) |
| Down syndrome | Hypotonia; characteristic facial features; single palmar crease; AVSD [7]; karyotype trisomy 21 |
| DiGeorge syndrome (22q11.2 deletion) | Cardiac abnormalities (conotruncal: interrupted aortic arch, truncus arteriosus, TOF), abnormal facies (low-set posteriorly rotated ears, hypertelorism), thymic hypo/aplasia, cleft palate, hypocalcaemia [7] — mnemonic: CATCH-22 |
| Williams syndrome | Mental retardation; elfin facies (full cheeks, flat nasal bridge, anteverted nostrils, long philtrum, prominent lips); hypercalcaemia; supravalvular AS; peripheral pulmonary artery stenosis [7] |
| Fetal alcohol spectrum disorder | Smooth philtrum, thin upper lip, short palpebral fissures; microcephaly; growth restriction; maternal alcohol history |
| Non-syndromic cystic hygroma | Isolated lymphatic malformation without other dysmorphic features; normal karyotype |
| Feature | Turner (45,X) | Noonan | Klinefelter (47,XXY) | MRKH | CAIS (46,XY) |
|---|---|---|---|---|---|
| Sex | Female | Both | Male | Female | Phenotypic female |
| Karyotype | 45,X or variant | Normal | 47,XXY | 46,XX | 46,XY |
| Stature | Short [2] | Short | Tall [2] | Normal | Normal/tall |
| Cardiac | Left-sided [7] | Right-sided [7] | MVP | Normal | Normal |
| Gonads | Streak ovaries | Usually normal | Small testes | Normal ovaries | Testes (inguinal/abdominal) |
| Puberty | Absent/delayed | Usually normal (may be delayed) | Delayed/incomplete | Normal | Breast development +, sparse hair |
| Intelligence | Normal, learning difficulties [2] | Mild ID in ~25% | Borderline-normal | Normal | Normal |
| Fertility | POI; IVF with donor ova [2] | Usually fertile | Infertility (azoospermia) [2] | Fertile (has ovaries) | Infertile |
The practical approach to a girl presenting with short stature ± delayed puberty:
- Auxology: Plot height on growth chart; calculate growth velocity; compare to mid-parental height
- Bone age: Delayed (CDGP, GHD, hypothyroidism) vs normal/near-normal (familial, TS)
- Bloods: CBP, ESR, LFT, RFT, electrolytes, bone profile, thyroid function, IGF-1 [6]
- Karyotype in girls [6] — mandatory in any girl with unexplained short stature (even if no dysmorphic features — mosaic TS can be cryptic)
- FSH/LH (if pubertal delay): ↑↑ = primary hypogonadism (TS, gonadal dysgenesis); ↓/normal = secondary (hypothalamic/pituitary)
- Provocative growth hormone testing [6] — if GHD suspected (low IGF-1, low growth velocity, delayed bone age)
- Pelvic USS: Assess for uterus and ovaries (absent in MRKH, CAIS; streak gonads in TS)
- Echocardiography and renal USS: Screen for associated structural anomalies if TS confirmed
Don't Get Caught Out
A common exam pitfall: Mosaic Turner syndrome (45,X/46,XX) can present with minimal or NO dysmorphic features — sometimes the only finding is unexplained short stature or primary ovarian insufficiency. This is why karyotype is recommended for ALL girls with unexplained short stature [6]. Do not wait for classic dysmorphic features before ordering a karyotype.
High Yield Summary – Differential Diagnosis of Turner Syndrome
- Short stature DDx in a girl: Familial short stature, CDGP, GHD, hypothyroidism, chronic disease, skeletal dysplasia, Noonan syndrome, isolated SHOX haploinsufficiency — always karyotype girls with unexplained short stature [6]
- Delayed puberty / primary amenorrhoea DDx: Organised by HPG axis level:
- Primary hypogonadism (↑FSH/LH): Turner, 46,XX/46,XY gonadal dysgenesis, autoimmune oophoritis, galactosaemia, chemo/radiation
- Secondary hypogonadism (↓FSH/LH): CDGP, Kallmann, functional hypothalamic, hyperprolactinaemia, hypopituitarism, iron overload
- Anatomical: MRKH, CAIS, imperforate hymen, transverse vaginal septum
- Noonan vs Turner: Both sexes vs females only; normal karyotype vs abnormal; right-sided cardiac (PS, HCM) vs left-sided cardiac (CoA, BAV) [7]
- Gonadal dysgenesis is the most common cause of primary amenorrhoea (43%) [8]
- Turner syndrome is primary hypogonadism → appropriately elevated FSH/LH [5]
- Y-material in Turner → risk of gonadoblastoma → prophylactic gonadectomy
- Isolated SHOX haploinsufficiency accounts for 2.2–4.2% of idiopathic short stature [2]
Active Recall – Differential Diagnosis of Turner Syndrome
References
[2] Senior notes: Adrian Lui Pediatrics Notes.pdf (p506–507, Turner Syndrome section) [5] Senior notes: Block A - I keep on bumping into people on my side_ pituitary tumours; hypopituitarism.pdf (Hypogonadism section) [6] Lecture slides: CFB (PAE02) Child growth and development.pdf (p56, Investigations for short stature) [7] Senior notes: Ryan Ho Cardiology.pdf (p185, Common syndromes associated with congenital heart diseases table) [8] Senior notes: Adrian Lui Gynecology Notes.pdf (p26, Approach to Amenorrhea)
Diagnostic Criteria, Algorithm, and Investigations for Turner Syndrome (45,X)
Turner syndrome does not have a formal set of diagnostic criteria like, say, the Jones criteria for rheumatic fever. Instead, the diagnosis rests on two pillars:
- Clinical suspicion — based on characteristic phenotypic features at any age
- Cytogenetic confirmation — demonstrating a missing or structurally abnormal X chromosome
The diagnosis of Turner syndrome is confirmed by karyotype analysis showing complete or partial absence of one X chromosome in a phenotypic female. No minimum number of clinical features is required — the karyotype is definitive.
When Is Karyotyping Indicated?
The threshold for ordering a karyotype should be low in any girl with suggestive features. Specifically:
GC Lecture – High Yield Exam Point
"Karyotype in girls" is a mandatory investigation in the workup of short stature [6]. The CFB (PAE02) lecture states: "Female with complete or partial absence of one X chromosome may have features of Turner syndrome including short stature, broad chest, widely spaced nipples, webbed neck, increase in carrying angle, and other system disorders such as cardiac defects, renal anomalies and hypothyroidism" [6]. This reinforces: any unexplained short stature in a girl warrants karyotyping, even without classic dysmorphic features.
Indications for karyotyping include:
- Unexplained short stature in any girl
- Delayed or absent puberty / primary amenorrhoea
- Classic dysmorphic features (webbed neck, lymphoedema, shield chest, cubitus valgus)
- Antenatal findings (cystic hygroma, hydrops, increased nuchal translucency, left-sided cardiac lesion on fetal echo)
- Neonatal lymphoedema of hands and feet
- Left-sided congenital heart disease (especially CoA or BAV) in a female infant
- Elevated FSH in a girl or adolescent (indicating primary ovarian insufficiency)
The algorithm below covers the three main clinical entry points — antenatal, neonatal/childhood, and adolescent — and the stepwise approach to confirmation and baseline screening once the diagnosis is established.
III. Investigation Modalities — Detailed
A. Cytogenetic / Genetic Investigations (Diagnostic)
Karyotyping is usually sufficient to establish the diagnosis in most cases [3].
| Aspect | Detail |
|---|---|
| Sample | Peripheral blood (postnatal); amniotic fluid or chorionic villi (prenatal) |
| Method | G-banding of metaphase chromosomes from cultured lymphocytes; typically 20–30 cells analysed |
| Expected findings | 45,X (~50%); mosaic patterns (45,X/46,XX, 45,X/47,XXX, 45,X/46,XY); structural X abnormalities (isochromosome Xq, Xp deletion, ring X) |
Why might the initial karyotype be falsely normal?
REPEAT karyotyping if the initial test is normal in a patient with strong clinical suspicion of Turner's syndrome, by using a different tissue sample such as skin, buccal mucosa cell, or bladder epithelial cells in urine sample [3].
False negatives can be attributed to abnormal cell lines dying out with age in bone marrow, leaving a normal cell line in peripheral blood. Mosaic karyotypes may be distributed differently among tissues in the same patient [3].
This is a critical concept: in tissue-limited mosaicism, the 45,X cell line may be present in the gonads and other tissues but absent or present in very low proportions in the blood. Using a different tissue source (e.g., skin fibroblast culture) increases diagnostic yield.
Must Know for Exams
A normal blood karyotype does NOT exclude Turner syndrome if clinical suspicion is high. Repeat karyotyping on a different tissue (skin biopsy for fibroblast culture, buccal mucosa, or urine bladder epithelial cells) [3]. This is commonly tested.
Prenatal karyotyping with amniocentesis or chorionic-villi sampling (CVS) [3].
- No screening programme is available for Turner syndrome [3] — it is not part of routine aneuploidy screening (unlike trisomy 21/18/13)
- Prenatal diagnosis is indicated when routine USG scan shows characteristic features including cystic hygroma and renal anomalies [3]
- Postnatal karyotyping with peripheral blood sample is recommended for confirmation of prenatal testing or for girls with typical signs of Turner syndrome [3]
Why confirm prenatally diagnosed TS postnatally? Because (a) confined placental mosaicism can give false-positive prenatal results, and (b) the proportion of 45,X cells in different tissues may differ, affecting prognosis and management.
Non-invasive prenatal testing (NIPT) using cell-free fetal DNA can now detect sex chromosome aneuploidies with reasonable sensitivity, but karyotype remains the gold standard for confirmation.
Fluorescent-in-situ-hybridisation (FISH) — indicated to look for occult Y chromosome mosaicism [3].
- Presence of Y chromosome material is associated with an increased risk of gonadoblastoma [3]
- Should be performed in all patients with Turner syndrome (some centres use PCR for SRY gene as an alternative or adjunct)
- If Y-material is found → prophylactic gonadectomy (lifetime gonadoblastoma risk ~15–30% in dysgenetic gonads with Y-material)
- Not always required but increasingly used in conjunction with standard karyotyping
- Can detect small deletions/duplications not visible on G-banding
- Particularly useful for characterising marker chromosomes and ring chromosomes
- Can identify cryptic Y-chromosome material
B. Endocrine Investigations
| Parameter | Expected Finding in Turner Syndrome | Interpretation |
|---|---|---|
| FSH | Elevated (often markedly, > 25–40 IU/L) | Reflects primary gonadal failure — the pituitary detects low oestrogen and increases FSH output |
| LH | Elevated | Same mechanism as FSH |
| Oestradiol | Low / undetectable | Streak gonads produce no/minimal oestrogen |
This is primary hypogonadism (hypergonadotrophic hypogonadism) — low oestrogen with appropriately high FSH/LH [5][9].
Age-specific interpretation (paediatric nuance):
- In infancy (mini-puberty): FSH/LH physiologically elevated in first 1–2 years → may see elevated gonadotrophins in TS infants (useful early diagnostic window)
- In mid-childhood (age 2–8): The HPG axis is quiescent → FSH/LH may be normal even in TS → less useful diagnostically at this age
- In pubertal age (> 10 years): FSH/LH should be rising as puberty activates → failure to see oestrogen rise with elevated gonadotrophins confirms ovarian failure
Confirm diagnosis of primary ovarian insufficiency: FSH > 25 IU/L × 2, ≥ 4 weeks apart [10].
Hormonal Profile Summary – Hypergonadotrophic Hypogonadism
Hypergonadotropic hypogonadism (Class 3) [10]:
- Hormonal profile: ↑FSH/LH, ↓E2 [10]
- Pelvic US (in primary amenorrhoea): uterus may be atrophic; may be hard to locate ovaries [10]
- Withdrawal bleeding test: negative for progestogen; positive with OCP → i.e., functioning uterus but no endogenous oestrogen [10]
This pattern is the classic endocrine fingerprint of Turner syndrome in the adolescent/adult.
TFT + Thyroid autoantibodies (Anti-TPO, Anti-TSH receptor) [3].
- Hypothyroidism occurs in 25–30% of TS patients, due to Hashimoto thyroiditis [2]
- Baseline TFT at diagnosis, then annual screening is recommended
- Expected findings in Hashimoto: ↑TSH, ↓/normal free T4, positive anti-TPO antibodies
- Why screen annually? Autoimmune thyroiditis can develop at any age and is insidious
- Diabetes mellitus risk is 2–4× increased [2]
- Screen with fasting glucose ± HbA1c at diagnosis and periodically
- OGTT if impaired fasting glucose is detected
- Both T1DM (autoimmune) and T2DM (insulin resistance) occur in TS
- IGF-1 is part of the standard short stature workup [6]
- In Turner syndrome, IGF-1 may be normal or low — the short stature is primarily due to SHOX haploinsufficiency rather than GH deficiency
- However, if IGF-1 is markedly low, provocative GH testing [6] may be indicated to exclude coexistent GH deficiency
- In practice, GH treatment is given for TS regardless of GH status (it is an approved indication for GH therapy), but GH deficiency should still be assessed
- Markers of ovarian reserve
- In classic 45,X: AMH and inhibin B are usually very low or undetectable, confirming minimal/no ovarian follicular reserve
- In mosaic TS (45,X/46,XX): may be detectable → better prognosis for spontaneous puberty and potential fertility
- Useful for fertility counselling in adolescents
| Test | Rationale |
|---|---|
| Lipid profile | Increased cardiovascular risk; dyslipidaemia screening |
| Liver function tests | LFT [3] — elevated transaminases are common in TS (hepatic steatosis, fibrosis); baseline and periodic monitoring |
C. Cardiac Investigations
Cardiovascular disease is the leading cause of morbidity and early mortality in Turner syndrome. Systematic cardiac evaluation is essential.
ECG / Transthoracic echocardiography / Cardiac MRI [3].
| Finding | Significance |
|---|---|
| Bicuspid aortic valve (BAV) | 10–15% [2]; may be functionally normal initially but predisposes to AS, AR, and aortic root dilatation over time |
| Coarctation of the aorta (CoA) | ~10% [2]; discrete narrowing of descending aorta at insertion of ductus [4]; look for narrowing with turbulent flow on Doppler |
| Aortic root dilatation | 8–28% [2]; must be indexed to body surface area (BSA) in paediatrics — use aortic size index (ASI) |
| Mitral valve prolapse | Can occur |
| Hypoplastic left heart syndrome | Rare but described [7] |
- Timing: At diagnosis → then every 3–5 years if initial echo normal; more frequently if abnormalities detected
- In neonates with suspected TS + cardiovascular collapse: look for duct-dependent CoA with collapse, shock, oliguria after ductal closure [4]
- Gold standard for detailed aortic assessment in older children/adolescents (usually from age ~8–10 when cooperation allows)
- Superior to echo for visualising the aortic arch, isthmus, and ascending aorta
- Essential for:
- Measuring aortic dimensions accurately (indexed to BSA)
- Detecting elongation of the transverse aorta (an early sign of aortopathy)
- Monitoring aortic root dilatation serially
- Detecting partial anomalous pulmonary venous return (PAPVR) — more common in TS than appreciated
- Recommended every 5 years if initial cardiac MRI is normal; more frequently if dilatation detected
- Look for:
- Right axis deviation (in neonates with CoA)
- LVH pattern (in chronic CoA or hypertension)
- Prolonged QTc — TS is associated with QTc prolongation; screen and avoid QT-prolonging drugs
- Conduction abnormalities
Hypertension occurs in ~20% [2] and may be present even without CoA.
- Measure in all four limbs at diagnosis to detect CoA gradient (> 20 mmHg upper-to-lower limb gradient is significant)
- Annual BP monitoring thereafter
- Use appropriate paediatric cuff sizes and compare to age/sex/height-specific normative tables
1. Renal Ultrasound (USS)
Renal ultrasound (USG) [3].
| Finding | Frequency | Significance |
|---|---|---|
| Horseshoe kidney | ~10% | Usually asymptomatic; may predispose to UTI, obstruction |
| Duplex collecting system | Variable | Risk of vesicoureteric reflux |
| Renal malrotation | Variable | Usually incidental |
| Absent/ectopic kidney | Rare | Assess total renal function |
| Hydronephrosis | Secondary | May indicate obstruction |
- Timing: At diagnosis; typically once unless abnormalities found
- RFT [3] and urinalysis [3] to assess renal function
- At diagnosis and then every 1–3 years throughout childhood and adolescence
- Types of hearing loss in TS:
- Conductive: Recurrent otitis media → middle ear effusion (from eustachian tube dysfunction due to craniofacial anatomy); most common in childhood
- Sensorineural: Progressive, particularly mid-frequency dip; becomes significant in adolescence/adulthood (X-linked inner ear gene effects)
- Recurrent otitis media / hearing loss affects ~60% [2]
- Early detection → hearing aids, speech therapy, grommets for recurrent OME as needed
F. Skeletal / Radiological Investigations
- Part of standard short stature workup [6]
- In Turner syndrome: bone age is usually mildly delayed relative to chronological age (typically 1–3 years behind)
- Helps predict adult height and guide timing of GH therapy and oestrogen replacement
- For baseline bone mineral density assessment
- Indicated in adolescence or when oestrogen replacement is being considered
- Turner syndrome predisposes to osteoporosis due to oestrogen deficiency (POI) — oestrogen is critical for bone mineralisation
- DXA scan for baseline BMD [10]
- Not routine, but if Madelung deformity, scoliosis, or other skeletal anomalies are suspected on clinical exam
- Look for: shortened 4th metacarpal on hand X-ray, Madelung deformity on wrist films, scoliosis on spine films
| Investigation | Rationale |
|---|---|
| LFT [3] | Elevated transaminases common; screen for hepatic steatosis/fibrosis |
| RFT [3] | Baseline renal function |
| Urinalysis [3] | Screen for renal anomaly-related complications |
| Serum blood glucose [3] | Screen for DM |
| Coeliac screen (anti-tTG IgA + total IgA) | ~6% prevalence of coeliac disease in TS; screen at diagnosis and every 2–5 years |
| Lipid profile | Cardiovascular risk assessment |
| 25-OH Vitamin D | Osteoporosis risk; guide supplementation |
| Ophthalmology review | Myopia, strabismus, amblyopia; baseline and periodic |
| Developmental/educational assessment | Learning difficulties with normal intelligence [2]; visuospatial, maths, executive function, social cognition |
| Investigation | At Diagnosis | Annual | Every 3–5 Years | As Indicated |
|---|---|---|---|---|
| Karyotype (blood ± FISH for Y) | ✓ | Repeat on different tissue if FN | ||
| Echocardiography | ✓ | ✓ (if normal) | More often if abnormal | |
| Cardiac MRI | From ~age 8–10 | ✓ (every 5y if normal) | More often if aortic dilatation | |
| BP (all four limbs at dx) | ✓ | ✓ | ||
| Renal USS | ✓ | If abnormalities found | ||
| TFT + anti-TPO | ✓ | ✓ | ||
| FSH, LH, oestradiol | ✓ (at pubertal age) | ✓ | Before puberty induction | |
| Fasting glucose / HbA1c | ✓ | ✓ | If risk factors | |
| LFT | ✓ | ✓ | ||
| Coeliac screen | ✓ | Every 2–5y | If symptomatic | |
| Lipid profile | ✓ | ✓ | ||
| Audiometry | ✓ | Every 1–3y | If hearing concerns | |
| Bone age X-ray | ✓ | Before GH therapy | ||
| DXA | Adolescence | ✓ | Before/during oestrogen Tx | |
| Eye examination | ✓ | ✓ | If visual concerns | |
| Developmental assessment | ✓ | If learning difficulties |
Common Exam Mistakes
- Forgetting to karyotype girls with short stature — Mosaic TS can have minimal dysmorphism; karyotype is indicated in all girls with unexplained short stature [6]
- Accepting a normal blood karyotype as ruling out TS — False negatives occur because abnormal cell lines may die out in bone marrow; repeat on skin/buccal mucosa/urine if suspicion is high [3]
- Not checking for Y-material — Missing Y mosaicism = missing gonadoblastoma risk = potential malignancy
- Measuring BP in upper limbs only — Miss CoA! Always measure in all four limbs at diagnosis
- Confusing primary and secondary hypogonadism — In TS, FSH/LH are high (hypergonadotropic hypogonadism) [9], not low
- Forgetting autoimmune screening — Hashimoto thyroiditis, coeliac disease, and T1DM are common comorbidities requiring ongoing surveillance
High Yield Summary – Diagnosis of Turner Syndrome
- Definitive diagnosis: Karyotype showing 45,X or variant (mosaic, structural X abnormality)
- Karyotype indicated in all girls with unexplained short stature [6]
- No prenatal screening programme exists for TS; prenatal karyotyping indicated when USS shows cystic hygroma, renal anomalies [3]
- If blood karyotype normal but clinical suspicion high → repeat on different tissue (skin, buccal, urine) [3]
- FISH for Y-chromosome material in all TS patients → if positive, prophylactic gonadectomy [3]
- Endocrine pattern: ↑FSH/LH, ↓oestradiol = hypergonadotropic hypogonadism = primary ovarian failure [5][9][10]
- Confirm POI: FSH > 25 IU/L × 2, ≥ 4 weeks apart [10]
- Baseline screening at diagnosis: Echo, renal USS, TFT + anti-TPO, LFT, RFT, glucose, coeliac screen, audiometry, bone age, ophthalmology, developmental assessment
- Ongoing surveillance: Annual TFT, BP, LFT; periodic echo/cardiac MRI, audiometry, coeliac screen, DXA, lipids, glucose
Active Recall – Diagnosis of Turner Syndrome
References
[2] Senior notes: Adrian Lui Pediatrics Notes.pdf (p506–507, Turner Syndrome section) [3] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (p847–850, Turner Syndrome: Etiology and Diagnosis sections) [4] Senior notes: Ryan Ho Cardiology.pdf (p190, Coarctation of Aorta section) [5] Senior notes: Block A - I keep on bumping into people on my side_ pituitary tumours; hypopituitarism.pdf (Hypogonadism section) [6] Lecture slides: CFB (PAE02) Child growth and development.pdf (p56, Investigations for short stature) [7] Senior notes: Ryan Ho Cardiology.pdf (p185, Common syndromes associated with congenital heart diseases table) [9] Senior notes: Block A - Introduction to Endocrine investigations.pdf (p1, Hypogonadism classification) [10] Senior notes: Adrian Lui Gynecology Notes.pdf (p29, Hypergonadotropic hypogonadism workup and investigation findings)
Management of Turner Syndrome (45,X)
Turner syndrome is a lifelong, multi-system condition — there is no cure. Management is therefore:
- Multidisciplinary — paediatric endocrinologist (lead), cardiologist, nephrologist, audiologist, psychologist, geneticist, gynaecologist (in adolescence), ophthalmologist, orthopaedic surgeon as needed
- Longitudinal — from diagnosis through transition to adult care
- Family-centred — with age-appropriate communication to the child and full involvement of caregivers; discuss diagnosis, prognosis, and treatment options openly; provide psychological support
- Phase-dependent — priorities shift with age (growth optimisation in childhood → puberty induction in adolescence → cardiovascular surveillance and fertility counselling into adulthood)
GC Lecture – High Yield Exam Point
III. Treatment Modalities — Detailed
A. Growth Hormone (GH) Therapy — Management of Short Stature
This is one of the two pillars of TS management (along with sex hormone replacement).
GH replacement for short stature [2].
- Short stature in TS is primarily due to SHOX gene haploinsufficiency [2], not GH deficiency
- However, supraphysiological doses of GH can still accelerate growth velocity and improve adult height by directly stimulating growth plate chondrocytes and increasing IGF-1
- GH is an approved indication for Turner syndrome in most jurisdictions (FDA, EMA) — it does not require demonstration of GH deficiency
| Aspect | Detail |
|---|---|
| Timing | Should start early, around 4–6 years of age, and preferably before 12–13 years old [3] |
| Dose | Requires higher dose of GH compared with standard dose used for GH deficiency [3]: typically 0.045–0.050 mg/kg/day (cf. GHD dose ~0.025–0.035 mg/kg/day) |
| Route | Subcutaneous injection, given daily (usually at bedtime, as this mimics physiological nocturnal GH secretion) |
| Expected outcome | Height can be normalised in most patients with Turner syndrome [3] — average gain of ~5–8 cm in adult height with optimal treatment |
| Monitoring | Growth velocity (should increase ≥ 2 cm/year above pre-treatment rate in first year); IGF-1 (keep within +2 SD for age); bone age annually; glucose homeostasis (GH can worsen insulin resistance); scoliosis monitoring (GH can accelerate pre-existing scoliosis) |
| Duration | Continue until growth velocity < 2 cm/year and/or bone age ≥ 14 years (epiphyseal fusion) |
- Growth failure in TS becomes evident by age 3–5 years. Starting GH at 4–6 years allows maximal duration of therapy before epiphyseal closure.
- Starting late (after age 12–13) gives insufficient time for meaningful height gain, especially since oestrogen (for puberty induction) will eventually promote epiphyseal fusion.
- The short stature is not due to GH deficiency — the growth plates are inherently less responsive due to SHOX haploinsufficiency. Higher doses of GH are needed to overcome this relative resistance and achieve clinically meaningful growth acceleration.
| Contraindication / Caution | Reason |
|---|---|
| Active malignancy | GH is mitogenic; avoid in active cancer |
| Uncontrolled diabetes | GH worsens insulin resistance → hyperglycaemia |
| Severe scoliosis | GH-mediated growth can worsen curvature |
| Closed epiphyses | No further growth potential |
| Active intracranial hypertension | GH can exacerbate pseudotumour cerebri (benign intracranial hypertension) — rare but monitor |
- Injection site reactions (local erythema, lipoatrophy) — rotate sites
- Fluid retention (rare in children, more common in adults)
- Benign intracranial hypertension (headache, papilloedema) — uncommon; reversible on stopping GH
- Slipped capital femoral epiphysis (SCFE) — rare; present with hip/knee pain and limp; requires urgent orthopaedic referral
- Insulin resistance → impaired glucose tolerance / T2DM — monitor fasting glucose annually (especially relevant in TS given baseline ↑DM risk)
Exam High Yield – GH Therapy in Turner Syndrome
B. Sex Hormone Replacement — Puberty Induction and Maintenance
Oestrogen replacement for gonadal insufficiency [2].
This is the second pillar of TS management. Since ovarian dysgenesis (95%) [2] leads to premature ovarian insufficiency (POI) [2], most girls with TS will require exogenous oestrogen to induce puberty and then ongoing combined oestrogen-progestogen replacement for bone health, cardiovascular protection, and well-being.
Low-dose oestrogen therapy should be started at 11–12 years old if there is no evidence of thelarche (breast development) and if FSH is high or AMH is low [3].
| Aspect | Detail |
|---|---|
| Timing | Age 11–12 years [3]; do not delay — this timing mimics normal pubertal onset and avoids psychosocial harm of delayed puberty |
| Route | Transdermal patch is preferred over oral administration [3] |
| Why transdermal? | More physiologic delivery into systemic rather than hepatic circulation (bypasses first-pass hepatic metabolism, avoids stimulation of production of coagulation factors and has lower risk of venous thromboembolism) [3] |
| Starting dose | Initially low [3] — e.g., low-dose transdermal 17β-oestradiol (start at 3.1–6.2 μg/day via quarter or half of a 25 μg patch, applied nocturnally or continuously) |
| Dose escalation | Gradual increase in dose so as to mimic normal puberty and breast development [3] — increase every 6 months over 2–3 years to full adult replacement dose (~50–100 μg/day transdermal, or 1–2 mg/day oral oestradiol) |
| Alternative route | Oral 17β-oestradiol (second line): start 0.25–0.5 mg/day, gradually increase. Ethinyl oestradiol is not recommended as it is synthetic, has excessive hepatic effects, and is less physiological |
Why low and slow? Starting oestrogen at too high a dose or increasing too rapidly: (a) causes premature epiphyseal fusion → compromises final adult height (especially if GH therapy is still ongoing); (b) leads to suboptimal breast development (rapid oestrogen exposure → areolar growth without proper glandular development → tubular breast shape). Low-dose, gradually increasing oestrogen mimics the normal 2–3 year tempo of puberty.
- In practice, there is a tension between starting oestrogen (which promotes epiphyseal fusion) and continuing GH (which maximises height).
- Current consensus: do not delay oestrogen beyond age 12 just to gain extra height — psychological well-being and bone health are equally important.
- The low starting dose of oestrogen has minimal impact on growth velocity initially, allowing GH to continue working.
Cyclic progestins are added later to the regimen to induce cyclic uterine bleeding and prevent endometrial hyperplasia [3].
| Aspect | Detail |
|---|---|
| Timing | Typically added after 2 years of oestrogen monotherapy, at 13–14 years old [3]; or when breakthrough bleeding occurs [11] |
| Rationale | Unopposed oestrogen stimulates endometrial proliferation → risk of endometrial hyperplasia and, eventually, endometrial carcinoma. Cyclic progestogen induces secretory transformation and shedding (withdrawal bleed), preventing hyperplasia |
| Why not add earlier? | Progestin can compromise ultimate breast growth, hence should not be added until there is substantial breast development [11] |
| Regimen | Medroxyprogesterone acetate 5–10 mg/day for 10–14 days per calendar month; or micronised progesterone 200 mg/day for 10–14 days; or combined OCP (once full adult dose reached and breast development is satisfactory) |
| Aspect | Detail |
|---|---|
| Duration | Lifelong (until natural menopause age ~50 years) — this is essentially HRT for POI |
| Benefits | Bone health (prevents osteoporosis), cardiovascular protection, uterine health, psychological well-being, sexual health |
| Monitoring | Annual: BP, lipids, LFT, bone density (DXA every 3–5 years); assess compliance |
Common Exam Mistake
Do not confuse puberty induction oestrogen with oral contraceptive pills. The goals and dosing are completely different. Puberty induction uses very low-dose 17β-oestradiol that is gradually increased to mimic physiology. OCPs contain synthetic ethinyl oestradiol at much higher doses and are not appropriate for puberty induction.
Pregnancy possible with IVF using donated ova [2].
| Option | Detail |
|---|---|
| Oocyte donation + IVF | Standard approach for TS women who wish to conceive; uses donor eggs fertilised in vitro and transferred to the TS patient's hormonally-prepared uterus. Success rates comparable to other IVF indications |
| Oocyte/embryo cryopreservation | Oocyte cryopreservation after controlled ovarian hyperstimulation is a possible fertility preservation option in young mosaic females [3] — for mosaic TS with some residual ovarian function, this can be offered before follicular depletion occurs |
| Spontaneous conception | Rare (~2–5% of mosaic TS patients may achieve spontaneous pregnancy); higher risk of miscarriage and chromosomal abnormalities in offspring |
| Preconception counselling | Chance of spontaneous conception decreases rapidly with age [3]; risk of miscarriage is high [3] |
| Cardiac risk assessment before pregnancy | Mandatory — pregnancy imposes significant haemodynamic stress; aortic dilatation/dissection risk increases during pregnancy. Cardiac MRI and echocardiography required pre-conception. Pregnancy may be contraindicated if significant aortic dilatation or prior dissection |
Must Know – Pregnancy Risk in Turner Syndrome
Pregnancy in TS carries a significantly elevated risk of aortic dissection (estimated ~2% risk per pregnancy). All TS women considering pregnancy must have thorough cardiac evaluation including cardiac MRI with measurement of aortic dimensions indexed to BSA. This is a life-threatening risk that must be communicated clearly.
| Aspect | Detail |
|---|---|
| Indication | Confirmed Y-chromosome material on FISH or molecular testing (e.g., 45,X/46,XY mosaicism) |
| Rationale | Dysgenetic gonads containing Y-material have ~15–30% lifetime risk of gonadoblastoma (which can progress to malignant germ cell tumours — dysgerminoma) |
| Procedure | Bilateral gonadectomy (laparoscopic preferred in paediatrics) |
| Timing | Once Y-material is confirmed; typically performed in childhood |
| Post-operative | Continue sex hormone replacement (as above) — same as for TS without Y-material |
E. Management of Specific Comorbidities
| Complication | Management |
|---|---|
| Coarctation of aorta (CoA) | Neonatal: prostaglandin E1 infusion to maintain ductus arteriosus if duct-dependent [4]; surgical repair (end-to-end anastomosis or subclavian flap) or balloon angioplasty ± stenting. Post-repair: lifelong BP monitoring (residual HTN common even after repair [4]) |
| Bicuspid aortic valve (BAV) | Surveillance echocardiography every 3–5 years (or more frequently if haemodynamically significant); surgical AVR if severe AS or AR develops |
| Aortic root dilatation | Serial cardiac MRI (every 5 years if normal, annually if dilated); β-blocker or ARB to reduce aortic wall stress; prophylactic aortic root replacement if aortic size index (ASI) > 2.5 cm/m² or rapid progression |
| Hypertension | Hypertension (20%) [2]; lifestyle measures; pharmacotherapy — ACE inhibitors or ARBs are first line (also have theoretical benefit on aortic wall remodelling); avoid oestrogen-related HTN by using transdermal route |
Hypothyroidism (25–30%) due to Hashimoto thyroiditis [2].
| Management | Detail |
|---|---|
| Levothyroxine replacement | If TSH elevated with positive anti-TPO; paediatric dosing: 2–4 μg/kg/day in young children, adjusted by TFT |
| Monitoring | Annual TFT + anti-TPO; even if initially normal, autoimmune thyroiditis can develop at any age |
DM risk 2–4× increased [2].
| Management | Detail |
|---|---|
| Screening | Fasting glucose ± HbA1c at diagnosis and periodically |
| Lifestyle | Diet, exercise, weight management |
| Pharmacotherapy | As per standard paediatric/adult DM guidelines if diagnosed |
Recurrent otitis media ± hearing loss (60%) [2].
| Type | Management |
|---|---|
| Conductive (OME) | Grommets (ventilation tubes) for chronic/recurrent OME; hearing aids if persistent |
| Sensorineural (progressive) | Hearing aids; speech therapy; annual audiometry monitoring |
| Issue | Management |
|---|---|
| Scoliosis (11%) | Physiotherapy; bracing if progressive; surgical correction if severe |
| Osteoporosis | Adequate oestrogen replacement; calcium + vitamin D supplementation; weight-bearing exercise; DXA monitoring |
| Madelung deformity | Usually managed conservatively; surgical correction (Vickers procedure) if functionally limiting |
| Issue | Management |
|---|---|
| Horseshoe kidney / anomalies | Nephrology follow-up; monitor for UTI, obstruction; RFT monitoring |
| Management | Detail |
|---|---|
| Screening | Anti-tTG IgA + total IgA at diagnosis, then every 2–5 years |
| Treatment | Gluten-free diet if confirmed |
| Issue | Management |
|---|---|
| Myopia, strabismus | Ophthalmology review; corrective lenses; patching/surgery for amblyopia risk |
| Issue | Management |
|---|---|
| Learning difficulties with normal intelligence [2] | Educational psychology assessment; targeted support for visuospatial processing, maths, executive function |
| Body image / self-esteem | Psychological support; peer support groups (Turner syndrome associations) |
| Social cognition difficulties | Social skills training if needed |
Genetic counselling: short/long-term implications, recurrence risk, antenatal diagnosis in future pregnancy [2].
| Topic | Key Points |
|---|---|
| Recurrence risk | Low recurrence risk [2] — most cases are de novo |
| Family implications | Explain the chromosomal basis; reassure that it was not caused by anything the parents did |
| Future pregnancies | Offer prenatal karyotyping (amniocentesis/CVS) in subsequent pregnancies if desired, though risk is low |
| Transition | Discuss long-term implications: fertility, cardiovascular surveillance, lifelong hormone replacement |
- Structured transition programme from paediatric to adult services (typically at age 16–18 years in Hong Kong)
- Ensure continuity of:
- Endocrine follow-up (oestrogen replacement, thyroid monitoring, DM screening)
- Cardiology follow-up (aortic surveillance — cardiac MRI, BP monitoring)
- Audiology
- Bone health (DXA, calcium/vitamin D)
- Fertility counselling and reproductive planning
- Psychosocial support
| Age | Key Management Actions |
|---|---|
| Neonate | Baseline screening (echo, renal USS, karyotype confirmation, FISH for Y); if CoA → prostaglandin E1, surgical repair; genetic counselling |
| Age 4–6 years | Start GH therapy [3]; monitor growth velocity, IGF-1, bone age annually |
| Age 11–12 years | Start low-dose oestrogen (transdermal preferred) if no thelarche + high FSH / low AMH [3]; continue GH |
| Age 13–14 years | Add cyclic progestins [3]; continue oestrogen escalation; continue GH until growth velocity < 2 cm/year |
| Adolescence | Fertility counselling; transition planning; DXA for bone health; ongoing cardiac, thyroid, metabolic, hearing surveillance |
| Lifelong | Oestrogen-progestogen replacement; cardiac MRI every 5 years; annual TFT, BP, LFT; audiometry; DM screening; psychosocial support |
Approach [2]:
- GH replacement for short stature [2]
- Oestrogen replacement for gonadal insufficiency [2]
- Regular review of child developmental and health [2]
- Screening for hearing impairment, hypothyroidism, cardiac/renal anomalies ± referral [2]
- Genetic counselling: short/long-term implications, recurrence risk, antenatal diagnosis in future pregnancy [2]
High Yield Summary – Management of Turner Syndrome
- Two pillars: GH therapy (for short stature) + oestrogen replacement (for ovarian failure)
- GH: start age 4–6 years; higher dose than for GHD; can normalise adult height [3]
- Oestrogen: start age 11–12 if no thelarche + high FSH/low AMH; transdermal preferred (avoids first-pass hepatic metabolism → lower VTE risk); low dose, gradually increase [3]
- Cyclic progestins: add after 2 years of oestrogen monotherapy (age ~13–14) or with breakthrough bleeding; do not add early — compromises breast development [3][11]
- Y-material → prophylactic gonadectomy (gonadoblastoma risk)
- Fertility: IVF with donated ova; oocyte cryopreservation in mosaic females [3]
- Cardiac surveillance: Echo, cardiac MRI (aortic dimensions), BP in 4 limbs; manage CoA, BAV, HTN, aortic dilatation
- Hypothyroidism (25–30%): Levothyroxine; annual TFT [2]
- DM (2–4× risk): Screen fasting glucose/HbA1c [2]
- Hearing loss (60%): Audiometry; grommets; hearing aids [2]
- Genetic counselling: low recurrence risk; antenatal diagnosis in future pregnancies [2]
- Psychosocial support: Normal intelligence; target learning support for visuospatial/maths; psychology; peer groups
- Transition to adult care at age 16–18 years — structured programme
Active Recall – Management of Turner Syndrome
References
[2] Senior notes: Adrian Lui Pediatrics Notes.pdf (p506–507, Turner Syndrome section) [3] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (p851, Turner Syndrome: Treatment section) [4] Senior notes: Ryan Ho Cardiology.pdf (p190, Coarctation of Aorta section) [6] Lecture slides: CFB (PAE02) Child growth and development.pdf (p56, Investigations for short stature) [7] Senior notes: Ryan Ho Cardiology.pdf (p185, Common syndromes associated with congenital heart diseases table) [11] Senior notes: Adrian Lui Pediatrics Notes.pdf (p75, Delayed Puberty – Management section)
Complications of Turner Syndrome (45,X)
Turner syndrome is a lifelong multi-system condition. Complications arise from the underlying haploinsufficiency of X-linked genes and affect virtually every organ system. The key concept is that many complications are not "acute events" but rather progressive, age-dependent processes that require anticipatory screening and longitudinal follow-up.
The complications can be conceptualised by system and by the age at which they typically manifest:
| Timing | Dominant Complications |
|---|---|
| Antenatal / Neonatal | Hydrops fetalis, cystic hygroma, lymphoedema, congenital heart disease (especially CoA) |
| Childhood | Growth failure, conductive hearing loss, learning difficulties, cardiac anomalies, renal anomalies |
| Adolescence | Absent puberty/amenorrhoea, psychosocial difficulties, osteoporosis begins |
| Adulthood | Cardiovascular (aortic dissection, HTN), infertility, metabolic (DM, dyslipidaemia), osteoporosis, progressive hearing loss, autoimmune disease, hepatic disease |
II. Cardiovascular Complications (Leading Cause of Morbidity and Premature Mortality)
Cardiovascular disease is responsible for the majority of excess mortality in Turner syndrome. Life expectancy is reduced by approximately 10–13 years compared to the general female population, primarily due to cardiovascular causes.
| Feature | Detail |
|---|---|
| Aortic root dilatation | 8–28% of TS patients [2]; due to intrinsic aortopathy (cystic medial necrosis — similar to Marfan syndrome but from X-linked vascular gene haploinsufficiency) |
| Aortic dissection | 2.5% [2]; the most feared cardiovascular complication — can be fatal |
| Risk factors for dissection | BAV, CoA (repaired or unrepaired), hypertension, aortic root dilatation, pregnancy |
| Mechanism | Haploinsufficiency of X-linked genes involved in extracellular matrix maintenance → weakened aortic wall (cystic medial necrosis) → progressive dilatation under haemodynamic stress → intimal tear → dissection |
| Associated cardiac lesions | Idiopathic aortic dilatation → dissection, aneurysm [7] |
| Screening | Echocardiography at diagnosis; cardiac MRI from ~age 8–10 then every 5 years (more frequently if dilatation present); aortic size indexed to BSA (aortic size index, ASI) |
| Management | β-blockers or ARBs (to reduce aortic wall stress and slow dilatation rate); prophylactic aortic root replacement if ASI > 2.5 cm/m² or rapid progression (> 3 mm/year) |
| Pregnancy risk | ~2% risk of aortic dissection per pregnancy — mandatory pre-conception cardiac MRI; may be an absolute contraindication to pregnancy if significant dilatation |
Why does Turner syndrome cause aortopathy? The aortic wall requires normal extracellular matrix (collagen, elastin, fibrillin) for structural integrity. X-linked genes contribute to vascular smooth muscle function and matrix synthesis. Haploinsufficiency → subtle matrix disorganisation → the aorta is "weaker" and dilates under normal haemodynamic load. This process is accelerated by coexisting BAV (which creates turbulent flow → wall stress) and hypertension.
Must Know for Exams
Aortic dissection is the number one cause of sudden death in Turner syndrome. Risk factors to remember: BAV + CoA + HTN + aortic root dilatation + pregnancy. This is commonly tested.
Congenital heart defects affect ~20% of TS patients [7]. These are present from birth but may have progressive complications over the lifespan.
| Cardiac Lesion | Frequency | Complications |
|---|---|---|
| Bicuspid aortic valve (BAV) | 10–15% [2] | Progressive aortic stenosis (calcification of abnormal valve over decades) → LV pressure overload → LVH → heart failure; progressive aortic regurgitation → LV volume overload; infective endocarditis (turbulent flow over abnormal valve → endocardial damage → bacterial seeding); associated with accelerated aortic root dilatation |
| Coarctation of the aorta (CoA) | ~10% [2] | Neonatal: duct-dependent circulation → HF with shock if duct closes; Post-repair: residual/recurrent hypertension (even after successful repair, HTN persists in ~30–50% due to permanent alteration of arterial mechanics); re-coarctation (especially after balloon angioplasty); aortic aneurysm at repair site; berry aneurysm (associated with CoA → risk of intracranial haemorrhage) |
| Aortic stenosis (AS) | Variable [2] | Secondary to BAV; progressive LV outflow obstruction → exertional syncope, angina, HF |
| Mitral valve prolapse (MVP) | Described [7][12] | Usually asymptomatic; complications include progressive severe MR requiring MVR; emboli; atrial fibrillation [12] |
| Hypoplastic left heart syndrome | Rare [7] | Severe duct-dependent lesion requiring staged surgical palliation (Norwood procedure) or heart transplantation |
Hypertension (20%) [2].
| Aspect | Detail |
|---|---|
| Prevalence | ~20% in childhood; increases with age (up to 40–50% in adult TS) |
| Mechanisms | (1) Secondary to CoA (repaired or unrepaired); (2) Renal anomalies → renovascular or parenchymal HTN; (3) Intrinsic arterial stiffness from vascular gene haploinsufficiency; (4) Essential HTN (increased frequency) |
| Consequences | Accelerates aortic root dilatation → increases dissection risk; LVH; cerebrovascular disease; renal damage |
| Management | Regular monitoring (annual BP, 4-limb at diagnosis); lifestyle measures; ACE inhibitors/ARBs first line |
- Prolonged QTc interval — reported in ~25% of TS patients; mechanism unclear but thought to relate to ion channel gene expression affected by X-linked regulatory genes
- Clinical significance: increased risk of arrhythmia; avoid QT-prolonging medications (macrolides, certain antihistamines, antipsychotics)
- Screen with ECG at diagnosis and periodically
III. Endocrine Complications
Ovarian dysgenesis (95%) → premature ovarian insufficiency [2].
| Consequence | Detail |
|---|---|
| Absent puberty / primary amenorrhoea | No spontaneous breast development, no menarche; psychosocial impact |
| Infertility | Pregnancy possible with IVF using donated ova [2]; ~2–5% of mosaic patients may conceive spontaneously but with high miscarriage risk and risk of chromosomally abnormal offspring |
| Oestrogen deficiency effects | Osteoporosis (↓bone mineralisation), cardiovascular risk (loss of oestrogen's vasculoprotective effects), urogenital atrophy, impaired sexual health, psychological effects |
Why does oestrogen deficiency cause osteoporosis? Oestrogen normally suppresses osteoclast activity (by promoting osteoclast apoptosis and inhibiting RANKL). Without oestrogen → unopposed osteoclast-mediated bone resorption → net bone loss → osteoporosis → fragility fractures.
Hypothyroidism (25–30%) due to Hashimoto thyroiditis [2].
| Aspect | Detail |
|---|---|
| Mechanism | Autoimmune predisposition from X-linked immune regulatory gene haploinsufficiency → lymphocytic infiltration and destruction of thyroid tissue → progressive thyroid failure |
| Complications if untreated | Growth retardation (exacerbates already short stature), fatigue, weight gain, constipation, cold intolerance, cognitive slowing, goitre, myxoedema |
| Monitoring | Annual TFT + anti-TPO; some patients develop subclinical hypothyroidism first (↑TSH, normal fT4) before overt disease |
Diabetes mellitus (2–4× increased risk) [2].
| Type | Mechanism |
|---|---|
| Type 1 DM | Autoimmune predisposition (same basis as Hashimoto); autoimmune β-cell destruction |
| Type 2 DM | Increased insulin resistance (from altered body composition — increased truncal adiposity + decreased lean mass); metabolic syndrome association; GH therapy can exacerbate insulin resistance |
| Complications | Same as general DM population — microvascular (retinopathy, nephropathy, neuropathy) and macrovascular (CVD) complications; additive cardiovascular risk in TS which already has intrinsic vascular vulnerability |
- Insulin resistance, dyslipidaemia, central obesity — common even in childhood/adolescence
- Metabolic syndrome prevalence is higher than in the general population
- Increases cardiovascular risk — multiplicative with pre-existing vascular anomalies and hypertension
IV. Skeletal Complications
| Aspect | Detail |
|---|---|
| Mechanism | POI → chronic oestrogen deficiency → increased osteoclast activity → progressive bone loss |
| Additional factors | SHOX haploinsufficiency affects bone microarchitecture; GH therapy may improve but does not fully normalise BMD; vitamin D deficiency common |
| Scoliosis | 11% [2]; may be exacerbated by GH-mediated growth acceleration |
| Monitoring | DXA from adolescence; calcium, vitamin D, oestrogen replacement |
| Prevention | Adequate oestrogen replacement (the most important intervention); calcium 1000–1200 mg/day + vitamin D 400–800 IU/day; weight-bearing exercise |
Madelung deformity of the wrist [2] — premature fusion of volar-ulnar distal radial physis → dorsal subluxation of distal ulna → bayonet deformity → limited wrist ROM, pain, cosmetic concern. Directly linked to SHOX haploinsufficiency.
Turner syndrome has a broad predisposition to autoimmunity — thought to be from haploinsufficiency of X-linked immune regulatory genes (e.g., FOXP3 and other immune modulators, though exact mechanisms remain under investigation).
| Autoimmune Condition | Approximate Frequency | Consequence |
|---|---|---|
| Hashimoto thyroiditis | 25–30% [2] | Hypothyroidism (see above) |
| Coeliac disease | ~6% | Malabsorption → growth failure (exacerbates short stature), iron deficiency anaemia, osteoporosis, abdominal symptoms |
| Type 1 DM | Increased | Insulin-dependent diabetes |
| Inflammatory bowel disease | Increased | Crohn disease / ulcerative colitis |
| Alopecia areata | Increased | Patchy hair loss |
| Vitiligo | Increased | Depigmentation |
Why are autoimmune diseases so common in TS? The X chromosome contains numerous genes involved in immune regulation and tolerance. Having only one functional copy → immune dysregulation → loss of self-tolerance → autoantibody production → organ-specific autoimmune disease. The specific pattern (thyroid >> coeliac >> DM1) reflects the same target organ susceptibility seen in other polyautoimmune conditions.
| Feature | Detail |
|---|---|
| Elevated liver enzymes | Common (up to 50–80% of adult TS patients have raised GGT, ALT, and/or AST) |
| Hepatic steatosis | Non-alcoholic fatty liver disease (NAFLD) — from metabolic syndrome and insulin resistance |
| Hepatic fibrosis / cirrhosis | Rare but can occur; mechanism includes vascular anomalies of hepatic vessels (architectural distortion, nodular regenerative hyperplasia) |
| Monitoring | Annual LFT; hepatic USS if persistently elevated transaminases; avoid hepatotoxic medications where possible |
Recurrent otitis media ± hearing loss (60%) [2].
| Type | Age | Mechanism | Consequence |
|---|---|---|---|
| Conductive hearing loss | Childhood | Craniofacial dysmorphism (short eustachian tubes, narrow nasopharynx) → eustachian tube dysfunction → recurrent acute otitis media and chronic otitis media with effusion (OME) → conductive loss | Speech and language delay if not addressed; may require grommets |
| Sensorineural hearing loss | Adolescence → adulthood (progressive) | X-linked cochlear gene haploinsufficiency → progressive degeneration of cochlear hair cells; classically a mid-frequency dip (unusual pattern — most SNHL affects high frequencies first) | Progressive hearing impairment; may require hearing aids; significant impact on quality of life |
The mid-frequency sensorineural dip pattern is somewhat unique to Turner syndrome and should raise suspicion in an adolescent girl with TS and progressive hearing difficulty. This is different from noise-induced or presbycusis-type high-frequency loss.
Renal anomalies: horseshoe kidney, kidney agenesis [2].
| Anomaly | Complication |
|---|---|
| Horseshoe kidney (~10%) | Predisposition to UTI (altered ureteric drainage), obstruction (at ureteropelvic junction due to crossing anomalous vessels), nephrolithiasis |
| Duplex collecting system | Vesicoureteric reflux → recurrent UTI → renal scarring |
| Renal agenesis | Solitary kidney → preserve renal function; avoid contact sports |
| Malrotation | Usually asymptomatic but may predispose to obstruction |
Myopia and strabismus [2].
| Issue | Detail |
|---|---|
| Myopia | Increased frequency; requires corrective lenses |
| Strabismus | Risk of amblyopia if not detected and treated early (patching, surgery) |
| Hyperopia | Also increased |
| Ptosis | Rare but described |
| Red-green colour blindness | X-linked; in 45,X, no second X to compensate → colour vision deficiency if the single X carries the defective gene (same incidence as in males, ~8%) |
Learning difficulties with normal intelligence [2].
| Domain | Detail |
|---|---|
| Visuospatial processing | Impaired spatial awareness, difficulty with maps, geometry, block design tasks |
| Mathematics | Dyscalculia-type difficulties; out of proportion to overall IQ |
| Executive function | Difficulty with planning, organisation, working memory |
| Social cognition | Difficulty reading facial expressions, interpreting social cues; increased social anxiety |
| Attention | Increased prevalence of ADHD |
| Verbal IQ | Typically normal or above average — reassure families that overall intelligence is not impaired |
| Psychosocial | Low self-esteem (short stature, delayed puberty relative to peers), social isolation, anxiety, depression |
Why the specific neurocognitive profile? The X chromosome carries genes important for brain development, particularly in regions subserving visuospatial and social cognition (parietal lobe, amygdala, prefrontal cortex). Haploinsufficiency of these genes → the characteristic pattern of preserved verbal skills but impaired non-verbal/spatial skills.
| Feature | Detail |
|---|---|
| Prevalence | ~10% of TS patients have Y-chromosome material [3] |
| Risk | 15–30% lifetime risk of gonadoblastoma in dysgenetic gonads containing Y-material |
| Malignant transformation | Gonadoblastoma itself is benign but can transform into dysgerminoma (malignant germ cell tumour) in ~60% of cases |
| Prevention | Prophylactic bilateral gonadectomy once Y-material is identified |
| Feature | Detail |
|---|---|
| Neonatal lymphoedema | Dorsum of hands and feet; usually resolves in infancy but may persist |
| Chronic lymphoedema | Can recur in adolescence/adulthood, particularly in lower limbs |
| Intestinal lymphangiectasia | Rare; protein-losing enteropathy → hypoalbuminaemia, peripheral oedema |
| Chylous effusions | Rare; chylothorax or chylous ascites from lymphatic malformation |
For the minority of TS women who achieve pregnancy (via donor oocyte IVF or rarely spontaneous conception):
| Complication | Mechanism |
|---|---|
| Aortic dissection (~2% per pregnancy) | Pregnancy → increased blood volume, cardiac output, and aortic wall stress → dissection in pre-existing aortopathy |
| Hypertension / pre-eclampsia | Baseline HTN tendency + pregnancy haemodynamic stress |
| Gestational DM | Baseline insulin resistance + GDM risk factors |
| Miscarriage | High rate (~30–40%); chromosomal abnormalities in offspring more common |
| Small pelvis / cephalopelvic disproportion | Short stature → increased Caesarean section rate |
| System | Key Complications |
|---|---|
| Cardiovascular | BAV (10–15%), CoA (~10%), aortic root dilatation (8–28%), dissection (2.5%), HTN (20%) [2]; MVP [7]; QTc prolongation |
| Endocrine | POI (95%), hypothyroidism (25–30%), DM (2–4×) [2]; metabolic syndrome |
| Skeletal | Osteoporosis, scoliosis (11%), Madelung deformity, cubitus valgus |
| Autoimmune | Hashimoto, coeliac, T1DM, IBD, alopecia areata |
| Hepatic | Elevated transaminases, NAFLD, fibrosis/cirrhosis |
| Audiological | Recurrent OM ± hearing loss (60%) [2]; progressive SNHL |
| Renal | Horseshoe kidney, duplex system, hydronephrosis |
| Ophthalmological | Myopia, strabismus |
| Neurocognitive | Visuospatial deficits, dyscalculia, social cognition difficulties, ADHD |
| Psychosocial | Low self-esteem, anxiety, depression |
| Lymphatic | Lymphoedema, rare chylous effusions |
| Gonadal | Gonadoblastoma (if Y-material present) |
| Pregnancy | Aortic dissection, pre-eclampsia, GDM, miscarriage |
High Yield Summary – Complications of Turner Syndrome
The exam-critical complications to remember:
- Aortic root dilatation (8–28%) and dissection (2.5%) [2] — #1 cause of sudden death; screen with echo + cardiac MRI; risk factors: BAV, CoA, HTN, pregnancy
- Congenital heart disease: BAV (10–15%), CoA (~10%), AS [2]; MVP [7][12]; HLHS (rare) [7]
- Hypertension (20%) [2] — multifactorial; accelerates aortopathy
- Primary ovarian insufficiency (95%) [2] → absent puberty, infertility, osteoporosis
- Hashimoto thyroiditis (25–30%) [2] → hypothyroidism
- Diabetes mellitus (2–4× risk) [2]
- Hearing loss (60%) [2] — conductive (childhood OME) + sensorineural (progressive mid-frequency dip)
- Osteoporosis — from chronic oestrogen deficiency
- Autoimmune cluster — thyroid, coeliac, T1DM, IBD
- Gonadoblastoma — if Y-material present (prophylactic gonadectomy required)
- Neurocognitive: visuospatial and maths difficulties with normal verbal IQ
- Pregnancy complications: aortic dissection (~2%), pre-eclampsia, miscarriage
Active Recall – Complications of Turner Syndrome
References
[2] Senior notes: Adrian Lui Pediatrics Notes.pdf (p506–507, Turner Syndrome section) [3] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (p847–851, Turner Syndrome: Etiology and Treatment sections) [7] Senior notes: Ryan Ho Cardiology.pdf (p185, Common syndromes associated with congenital heart diseases table) [12] Senior notes: Block A - Fever and a murmur_ Valvular heart diseases; Infective endocarditis.pdf (p15, Mitral valve prolapse section)
High Yield Summary
Turner Syndrome (45,X) — Key Points for Exams:
- Definition: Phenotypic female with complete or partial loss of one X chromosome; classic karyotype 45,X
- Pathophysiology: Haploinsufficiency of genes on the X chromosome that escape X-inactivation [1]
- Incidence: ~1/2,500 live-born females; > 95% of 45,X conceptions miscarry [2]
- Karyotypes: 45,X (~50%); mosaic (~30%); structural X abnormalities (~15%); 10% may have Y material → risk of gonadoblastoma [3]
- SHOX gene haploinsufficiency → short stature (> 95%) and skeletal anomalies (cubitus valgus, Madelung deformity, shortened 4th metacarpal) [2]
- Ovarian dysgenesis (95%) → streak gonads → POI → delayed/absent puberty, primary amenorrhoea, infertility [2]
- Lymphatic hypoplasia → neonatal lymphoedema, cystic hygroma, webbed neck
- Cardiac: bicuspid AV (10–15%), CoA (~10%), aortic root dilatation (8–28%), dissection (2.5%), HTN (20%) [2][4]
- Autoimmune: Hashimoto thyroiditis (25–30%); coeliac disease; DM (2–4× risk) [2]
- Primary hypogonadism: Low oestrogen + appropriately elevated FSH/LH (↔ secondary hypogonadism = low FSH/LH) [5]
- Learning difficulties with normal intelligence — visuospatial/maths deficits [2]
- Y-material screening: If found → prophylactic gonadectomy (gonadoblastoma risk)
- Low recurrence risk [2]
High Yield Summary – Differential Diagnosis of Turner Syndrome
- Short stature DDx in a girl: Familial short stature, CDGP, GHD, hypothyroidism, chronic disease, skeletal dysplasia, Noonan syndrome, isolated SHOX haploinsufficiency — always karyotype girls with unexplained short stature [6]
- Delayed puberty / primary amenorrhoea DDx: Organised by HPG axis level:
- Primary hypogonadism (↑FSH/LH): Turner, 46,XX/46,XY gonadal dysgenesis, autoimmune oophoritis, galactosaemia, chemo/radiation
- Secondary hypogonadism (↓FSH/LH): CDGP, Kallmann, functional hypothalamic, hyperprolactinaemia, hypopituitarism, iron overload
- Anatomical: MRKH, CAIS, imperforate hymen, transverse vaginal septum
- Noonan vs Turner: Both sexes vs females only; normal karyotype vs abnormal; right-sided cardiac (PS, HCM) vs left-sided cardiac (CoA, BAV) [7]
- Gonadal dysgenesis is the most common cause of primary amenorrhoea (43%) [8]
- Turner syndrome is primary hypogonadism → appropriately elevated FSH/LH [5]
- Y-material in Turner → risk of gonadoblastoma → prophylactic gonadectomy
- Isolated SHOX haploinsufficiency accounts for 2.2–4.2% of idiopathic short stature [2]
High Yield Summary – Diagnosis of Turner Syndrome
- Definitive diagnosis: Karyotype showing 45,X or variant (mosaic, structural X abnormality)
- Karyotype indicated in all girls with unexplained short stature [6]
- No prenatal screening programme exists for TS; prenatal karyotyping indicated when USS shows cystic hygroma, renal anomalies [3]
- If blood karyotype normal but clinical suspicion high → repeat on different tissue (skin, buccal, urine) [3]
- FISH for Y-chromosome material in all TS patients → if positive, prophylactic gonadectomy [3]
- Endocrine pattern: ↑FSH/LH, ↓oestradiol = hypergonadotropic hypogonadism = primary ovarian failure [5][9][10]
- Confirm POI: FSH > 25 IU/L × 2, ≥ 4 weeks apart [10]
- Baseline screening at diagnosis: Echo, renal USS, TFT + anti-TPO, LFT, RFT, glucose, coeliac screen, audiometry, bone age, ophthalmology, developmental assessment
- Ongoing surveillance: Annual TFT, BP, LFT; periodic echo/cardiac MRI, audiometry, coeliac screen, DXA, lipids, glucose
High Yield Summary – Management of Turner Syndrome
- Two pillars: GH therapy (for short stature) + oestrogen replacement (for ovarian failure)
- GH: start age 4–6 years; higher dose than for GHD; can normalise adult height [3]
- Oestrogen: start age 11–12 if no thelarche + high FSH/low AMH; transdermal preferred (avoids first-pass hepatic metabolism → lower VTE risk); low dose, gradually increase [3]
- Cyclic progestins: add after 2 years of oestrogen monotherapy (age ~13–14) or with breakthrough bleeding; do not add early — compromises breast development [3][11]
- Y-material → prophylactic gonadectomy (gonadoblastoma risk)
- Fertility: IVF with donated ova; oocyte cryopreservation in mosaic females [3]
- Cardiac surveillance: Echo, cardiac MRI (aortic dimensions), BP in 4 limbs; manage CoA, BAV, HTN, aortic dilatation
- Hypothyroidism (25–30%): Levothyroxine; annual TFT [2]
- DM (2–4× risk): Screen fasting glucose/HbA1c [2]
- Hearing loss (60%): Audiometry; grommets; hearing aids [2]
- Genetic counselling: low recurrence risk; antenatal diagnosis in future pregnancies [2]
- Psychosocial support: Normal intelligence; target learning support for visuospatial/maths; psychology; peer groups
- Transition to adult care at age 16–18 years — structured programme
High Yield Summary – Complications of Turner Syndrome
The exam-critical complications to remember:
- Aortic root dilatation (8–28%) and dissection (2.5%) [2] — #1 cause of sudden death; screen with echo + cardiac MRI; risk factors: BAV, CoA, HTN, pregnancy
- Congenital heart disease: BAV (10–15%), CoA (~10%), AS [2]; MVP [7][12]; HLHS (rare) [7]
- Hypertension (20%) [2] — multifactorial; accelerates aortopathy
- Primary ovarian insufficiency (95%) [2] → absent puberty, infertility, osteoporosis
- Hashimoto thyroiditis (25–30%) [2] → hypothyroidism
- Diabetes mellitus (2–4× risk) [2]
- Hearing loss (60%) [2] — conductive (childhood OME) + sensorineural (progressive mid-frequency dip)
- Osteoporosis — from chronic oestrogen deficiency
- Autoimmune cluster — thyroid, coeliac, T1DM, IBD
- Gonadoblastoma — if Y-material present (prophylactic gonadectomy required)
- Neurocognitive: visuospatial and maths difficulties with normal verbal IQ
- Pregnancy complications: aortic dissection (~2%), pre-eclampsia, miscarriage
Memory palace hooks for Turner Syndrome (45,X)
How to Use This Memory Palace
Each numbered symbol is a recall hook mapped back to this page's own notes. The Note concept column is the source of truth; the symbol logic explains why the visual cue should trigger that concept.
This first pass maps the symbols supplied for labels 1-68. Any additional labels visible in the image should be added only after they are tied back to the MBBSPedia note sections.
Source Alignment Guardrail
The supplied Sketchy map includes a few phrased teaching hooks that are broader than this page's current notes, especially NIPT performance and exact surveillance intervals. This table anchors those hooks to the article's current concepts: cytogenetic confirmation by karyotype, baseline multi-system screening, and ongoing surveillance.
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 1 | Turning windmill | Summary / Etiology | Turner syndrome is a phenotypic female condition caused by complete or partial loss of one X chromosome; classic karyotype is 45,X. | The turning windmill cues "Turner" and the broad phenotype spinning out from one missing X. |
| 2 | #1 foam finger | Etiology / Summary | Turner syndrome occurs in about 1 in 2,500 live-born females, while most 45,X conceptions miscarry. | The foam finger marks this as a high-yield sex-chromosome aneuploidy in females. |
| 3 | Single X blade | Etiology | Classic 45,X monosomy accounts for about 40-50% of cases and usually produces the most complete phenotype. | A single X-shaped blade cues one sex chromosome instead of two. |
| 4 | "Putter junction" sign | Etiology | Classic Turner syndrome is usually de novo; paternal non-disjunction accounts for many cases. | "Putter junction" sounds like non-disjunction, the chromosome-segregation error. |
| 5 | Mosaic "X" flower pot | Etiology | 45,X mosaicism is common and often milder; some patients have spontaneous puberty or fertility. | A mosaic-patterned pot cues mixed cell lines in the same patient. |
| 6 | Mosaic "Y" flower pot | Etiology / Dx / Mx | About 10-12% may have Y-chromosome material; screen for it because it raises gonadoblastoma risk. | The Y-marked mosaic pot cues a hidden Y-containing cell line. |
| 7 | Broken X blade | Etiology | Structural X abnormalities include isochromosome Xq, Xp deletion, ring X, and marker chromosomes. | A broken X blade cues a structurally abnormal X chromosome. |
| 8 | MisCARRIAGE | Etiology / Summary | More than 95% of 45,X conceptuses miscarry; only a small minority survive to term. | The car in "miscarriage" turns early fetal loss into a visual hook. |
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 9 | Pregnant person with ultrasound bullhorn | Dx / Etiology | Antenatal ultrasound findings such as increased nuchal translucency, cystic hygroma, hydrops, cardiac lesions, or renal anomalies should raise suspicion. | The ultrasound bullhorn announces prenatal warning signs. |
| 10 | Thick neck scarf | Dx / Etiology | Increased nuchal translucency and cystic hygroma reflect fetal lymphatic obstruction. | A thick scarf around the neck cues excess nuchal fluid. |
| 11 | "Restricted" sign | Etiology / Dx | Growth failure is central in Turner syndrome; prenatal growth restriction strengthens suspicion when paired with other markers. | "Restricted" cues restricted fetal growth. |
| 12 | Cystic putter | Etiology / Dx | Cystic hygroma is a classic antenatal lymphatic finding in Turner syndrome. | The cystic-looking putter cues a fluid-filled hygroma. |
| 13 | Hydrops doll | Etiology / Complications | Fetal hydrops can occur from severe lymphatic dysfunction. | The swollen doll cues hydrops fetalis. |
| 14 | Left heart hazard | Etiology / Dx | Left-sided cardiac lesions, especially coarctation and bicuspid aortic valve, are high-yield Turner associations. | The hazard sign on the left side points to left-heart obstruction. |
| 15 | Kidney hazard | Dx / Complications | Renal anomalies such as horseshoe kidney, duplex collecting system, or malrotation are associated with Turner syndrome. | The kidney warning sign flags renal involvement. |
| 16 | Short sleeve and pant leg | Etiology / Summary | SHOX haploinsufficiency causes short stature and skeletal anomalies. | Short clothing cues shortened long bones and later short stature. |
| 17 | "NIP" free DNA samples | Dx | NIPT can suggest sex-chromosome aneuploidy, but karyotype remains the diagnostic gold standard. | Nipping small samples cues cell-free DNA screening, not definitive diagnosis. |
| 18 | Corndog samples | Dx | Prenatal diagnostic testing can use chorionic villus sampling for karyotype. | "Corndog" cues chorionic villus sampling as a sampled tissue. |
| 19 | Amnio apple juice samples | Dx | Prenatal diagnostic testing can use amniocentesis for karyotype. | The amnio-colored drink cues amniotic fluid sampling. |
| 20 | Churro types | Dx | Karyotype analysis confirms Turner syndrome and defines 45,X, mosaic, or structural variants. | Churro "types" cues chromosome karyotypes. |
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 21 | Female cutout | Etiology / DDx | Turner syndrome affects phenotypic females; Noonan syndrome can mimic it in both sexes. | The female cutout anchors the sex-specific phenotype. |
| 22 | Edematous hands and feet | Etiology / Summary | Neonatal lymphoedema of hands and feet is a classic presentation. | Puffy hands and feet directly cue lymphatic hypoplasia. |
| 23 | Webbed hat | Etiology / Summary | Webbed neck results from resolved fetal cystic hygroma. | The webbed hat makes the neck webbing memorable. |
| 24 | Low ear beads | Etiology | Low-set ears are part of the Turner craniofacial phenotype. | Beads hanging low cue low-set ears. |
| 25 | High arched hat | Etiology | Narrow high-arched palate contributes to feeding and dental issues. | The high arch of the hat cues a high palate. |
| 26 | Small gnome chin | Etiology | Micrognathia / retrognathia is a craniofacial feature of Turner syndrome. | The small chin cues underdeveloped mandible. |
| 27 | Dysplastic nails | Etiology | Spoon-shaped or hypoplastic nails are associated with Turner syndrome. | Abnormal nails directly cue nail dysplasia. |
| 28 | Short 4th club | Etiology / Summary | Shortened 4th metacarpal is a SHOX-related skeletal sign. | The shortened club marks the shortened 4th ray. |
| 29 | Short gnome | Etiology / Summary | Short stature is the most consistent physical finding and reflects SHOX haploinsufficiency. | The short gnome cues the key growth phenotype. |
| 30 | Low hairline / neckline | Etiology / Summary | Low posterior hairline is a common craniofacial sign. | The low neckline cues posterior hairline sitting low. |
| 31 | Shield chest with wide nipple beads | Etiology / Summary | Turner syndrome can cause a broad shield chest with widely spaced nipples. | The shield and wide beads cue the chest shape and nipple spacing. |
| 32 | Valgus arms | Etiology / Summary | Cubitus valgus is an increased carrying angle of the elbow. | Outward-angled arms cue valgus alignment. |
| 33 | Order churro types now | Dx / DDx | Order a karyotype in girls with unexplained short stature, delayed puberty, suggestive dysmorphism, or concerning prenatal findings. | Ordering churro "types" cues ordering a karyotype. |
| 34 | Turner syndrome DDx sign | DDx | Noonan syndrome is the key Turner mimic; it has normal karyotype and right-sided cardiac lesions. | The DDx sign reminds you to separate Turner from Noonan. |
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 35 | "Go Team!" sign | Mx | Management is multidisciplinary and longitudinal. | The team cheer cues coordinated endocrine, cardiac, renal, hearing, genetic, and psychosocial care. |
| 36 | Counselor | Mx / Summary | Genetic counselling should cover the de novo basis, low recurrence risk, prognosis, and future pregnancy options. | The counselor cues early family-centred counselling. |
| 37 | "Community" sign | Mx | Support groups and peer/community resources help families after diagnosis. | The community sign cues linking families with Turner resources. |
| 38 | Heart putter with ultrasound bullhorn | Dx / Mx | Cardiac evaluation at diagnosis includes echocardiography, with cardiac MRI for aortic assessment when age-appropriate. | Heart plus ultrasound cues echo-based cardiac screening. |
| 39 | ECG skirt | Dx | ECG screens for conduction abnormalities such as prolonged QTc. | The ECG pattern on clothing cues rhythm assessment. |
| 40 | Four-point sweatbands | Dx / Mx | Measure blood pressure in all four limbs at diagnosis to look for coarctation gradients. | Four bands cue the four extremity BP check. |
| 41 | Bicuspid doors | Etiology / Complications | Bicuspid aortic valve is a common Turner cardiac lesion. | Two door leaflets cue a bicuspid valve. |
| 42 | Pinched arch | Etiology / Complications | Coarctation of the aorta is a high-yield left-sided lesion in Turner syndrome. | A pinched arch cues a narrowed aortic arch. |
| 43 | Kidney putter with ultrasound bullhorn | Dx / Mx | Renal ultrasound is done at diagnosis to detect structural renal anomalies. | Kidney plus ultrasound cues renal USS. |
| 44 | Gripping ureter handle | Dx / Complications | Duplex collecting systems and drainage problems can lead to reflux, obstruction, or UTIs. | The gripped ureter cues collecting-system irregularity. |
| 45 | Horseshoe wicket | Dx / Complications | Horseshoe kidney is a classic Turner renal anomaly. | The horseshoe wicket directly cues horseshoe kidney. |
| 46 | Thyroid bowtie, "T4" hat, and antibody putter | Dx / Mx | Screen with TFTs and thyroid autoantibodies because Hashimoto thyroiditis and hypothyroidism are common. | T4 plus antibodies cues autoimmune thyroid screening. |
| 47 | Growth chart grass | Dx / Mx | Plot height and growth velocity, preferably on Turner-specific growth charts. | The chart-like grass cues longitudinal growth monitoring. |
| 48 | "GROW" fertilizer | Mx | Recombinant growth hormone, started early, improves growth velocity and adult height. | Fertilizer makes the child "grow," cueing GH therapy. |
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 49 | Failing female flowers with "Flower Stim LigHt" | Etiology / Dx | Primary ovarian insufficiency causes hypergonadotropic hypogonadism: high FSH/LH with low oestrogen. | Failing flowers cue ovarian failure; the phrase hides FSH and LH. |
| 50 | Dirt streaks | Etiology / Summary | Ovarian dysgenesis produces streak gonads from accelerated oocyte loss. | Streaks in the dirt cue streak ovaries. |
| 51 | Crossed-out breasts and white skirt | Dx / Etiology | Most patients have absent or incomplete puberty and primary amenorrhoea; AMH and FSH help assess ovarian reserve. | Crossed breasts cue absent thelarche; the skirt cues absent menses. |
| 52 | "Estrogrow" and "Protect" fertilizer | Mx | Low-dose oestrogen starts around 11-12 years if puberty is absent; cyclic progestins are added later for endometrial protection. | Estrogrow cues oestrogen; Protect cues later progestin protection. |
| 53 | Gonad removal | Dx / Mx / Complications | Y-chromosome material requires prophylactic gonadectomy because of gonadoblastoma risk. | Removing the gonad cues cancer-risk prevention. |
| No. | Symbol | Source tab | Note concept | Etymology / symbol logic |
|---|---|---|---|---|
| 54 | Brain putter with "Learn to Golf" book | Etiology / Mx / Complications | Intelligence is usually normal, but visuospatial, maths, executive, attention, and social-cognition difficulties are increased. | The learning book cues targeted educational support rather than global intellectual disability. |
| 55 | T4 hat and thyroid bowtie | Dx / Mx | Thyroid function should be checked at diagnosis and then regularly because autoimmune thyroiditis can develop later. | The T4 hat repeats the ongoing thyroid surveillance hook. |
| 56 | "SEALiac" risk pond | Dx / Mx / Complications | Coeliac disease is more common; screen with anti-tTG IgA plus total IgA at diagnosis and periodically. | The seal cues "coeliac" and the autoimmune screening cluster. |
| 57 | Covering painful ears | Dx / Mx / Complications | Recurrent otitis media causes conductive hearing loss; progressive sensorineural hearing loss can also occur, so audiology follow-up is important. | Painful covered ears cue both ear infections and hearing loss. |
| 58 | Glasses with deviated eyeBALLS | Dx / Mx / Complications | Eye problems include myopia, hyperopia, strabismus, and amblyopia risk; ophthalmology review is needed. | Glasses cue refractive error; crossed eyes cue strabismus. |
| 59 | High-pressure steam | Complications / Mx | Hypertension is common and accelerates aortopathy; BP needs ongoing monitoring. | High-pressure steam cues high blood pressure. |
| 60 | Dissected club | Complications | Aortic root dilatation and aortic dissection are feared long-term cardiovascular complications. | A split club cues aortic dissection. |
| 61 | "Estrogrow" with heart | Mx / Complications | Oestrogen replacement supports bone and cardiovascular health, alongside cardiac monitoring, BP control, lipids, and lifestyle care. | Oestrogen next to the heart cues its protective role within broader risk management. |
| 62 | Overweight figure, lipid donut, and glucose candy | Complications / Dx | Turner syndrome increases risk of central obesity, dyslipidaemia, insulin resistance, and diabetes. | Weight, fat, and sugar props cue metabolic surveillance. |
| 63 | Curved spine club with skull and X-bones | Complications / Mx | Scoliosis and other skeletal issues should be monitored, especially during growth treatment. | The curved club cues spinal curvature. |
| 64 | Shifted end of putter | Etiology / Complications | Madelung deformity and genu valgum/varum are SHOX-related skeletal problems. | The shifted putter end cues bayonet-like wrist deformity and malalignment. |
| 65 | Cracked weak bone post | Complications / Mx | Turner syndrome predisposes to low bone mineral density and osteoporosis, mainly from oestrogen deficiency plus intrinsic bone effects. | A cracked post cues fragile bone. |
| 66 | Low D sign | Dx / Mx / Complications | Vitamin D and calcium support bone health alongside oestrogen and growth hormone therapy. | "Low D" cues vitamin D deficiency risk and supplementation. |
| 67 | Infertility tree | Mx / Complications | Fertility is reduced; options include donor-oocyte IVF and, in selected mosaic patients, early oocyte cryopreservation. | A barren tree cues reduced fertility and fertility-preservation counselling. |
| 68 | Mortality skull | Complications | Turner syndrome has excess premature mortality, mainly from cardiovascular disease such as aortopathy, hypertension, and coronary disease. | The skull cues the mortality risk that makes cardiac surveillance essential. |
Klinefelter Syndrome (47,xxy)
Klinefelter syndrome is a sex chromosome aneuploidy (47,XXY) in males that typically presents during adolescence with tall stature, small firm testes, gynecomastia, delayed or incomplete puberty, and later infertility due to primary hypogonadism.
Achondroplasia
Achondroplasia is the most common form of skeletal dysplasia in children, caused by a gain-of-function mutation in the FGFR3 gene that impairs endochondral ossification, resulting in rhizomelic short-limbed dwarfism typically evident at birth.