Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss Syndrome)
Eosinophilic granulomatosis with polyangiitis is an ANCA-associated small- and medium-vessel vasculitis characterized by asthma, peripheral eosinophilia, and extravascular eosinophilic granulomas affecting multiple organ systems.
Eosinophilic Granulomatosis with Polyangiitis (EGPA) — Churg-Strauss Syndrome
Eosinophilic Granulomatosis with Polyangiitis (EGPA), formerly known as Churg-Strauss syndrome, is a systemic necrotizing vasculitis predominantly affecting small-to-medium vessels (capillaries, venules, arterioles, small arteries, and veins). It is characterised by three cardinal features [1][2][3]:
- Eosinophil-rich tissue infiltration
- Necrotizing granulomatous inflammation, often involving the respiratory tract
- Association with asthma and peripheral blood eosinophilia
Let's break the name down:
- Eosinophilic → dominated by eosinophils (a type of granulocyte involved in allergy and parasitic defence)
- Granulomatosis → formation of granulomas (organized collections of macrophages/epithelioid cells trying to wall off a persistent insult)
- Polyangiitis → inflammation of multiple ("poly") blood vessels ("angiitis")
EGPA is classified as an ANCA-associated vasculitis (AAV) alongside Granulomatosis with Polyangiitis (GPA, Wegener's) and Microscopic Polyangiitis (MPA), although only 40–50% of EGPA patients are ANCA-positive — making it the least consistently ANCA-associated of the three [1][2].
EGPA is an ANCA-associated small vessel vasculitis characterised by eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract and associated with asthma and eosinophilia [1][3].
Key Distinguishing Feature
Asthma is almost ALWAYS present in EGPA. If your vasculitis patient has asthma + eosinophilia, think EGPA first. Neither GPA nor MPA is typically associated with asthma.
2. Epidemiology
- Rare: annual incidence approximately 0.5–6.8 per million population worldwide [4]
- Prevalence approximately 10–14 per million
- Among patients with asthma, the incidence rises to approximately 34–67 per million per year — this makes sense because asthma is virtually a prerequisite
- EGPA is the least common of the three AAVs (GPA > MPA > EGPA in most Western registries; in East Asia, MPA is more common than GPA)
- More data from Caucasian populations; however, EGPA is well-described in East Asian populations including Hong Kong/Chinese patients
- In Hong Kong, awareness has increased with more cases being identified through rheumatology and respiratory clinics at Queen Mary Hospital and Prince of Wales Hospital
- MPA tends to be more common than GPA in East Asian populations, but EGPA remains rare across all ethnicities
There is no single well-defined "cause" of EGPA; rather, it develops in genetically predisposed individuals with existing atopic/allergic backgrounds. Key risk factors and associations include:
| Risk Factor | Mechanism/Explanation |
|---|---|
| Pre-existing asthma | Nearly universal (> 90–95%); the allergic airway disease is the soil from which EGPA grows — Th2-driven immune dysregulation is the common thread |
| Atopic background | Allergic rhinitis, nasal polyposis, eczema — suggests a Th2-skewed immune phenotype |
| HLA associations | HLA-DRB104 and HLA-DRB107 have been reported, suggesting genetic susceptibility involving MHC class II molecules that influence antigen presentation |
| Leukotriene receptor antagonist (LTRA) use | Montelukast/zafirlukast may unmask previously undiagnosed Churg-Strauss Syndrome [5] — the proposed mechanism is that LTRA allows steroid dose reduction in asthma patients, and the reduced steroid burden "unmasks" the underlying vasculitic disease that was being suppressed by corticosteroids (see detailed discussion below) |
| Drug-related triggers | Macrolide antibiotics, carbamazepine, quinine (rare case reports) |
| Vaccination (rare reports) | Temporal association only; causality unproven |
LTRA and EGPA — High Yield Exam Point
Leukotriene receptor antagonists unmask previously undiagnosed Churg-Strauss Syndrome (or eosinophilic granulomatosis with polyangiitis) [5]. This is a classic exam vignette: a patient with "difficult asthma" is switched from inhaled corticosteroids to montelukast → systemic manifestations (vasculitis, eosinophilia, neuropathy) emerge. The LTRA itself doesn't cause EGPA — rather, the reduction in steroid dose that accompanies the switch removes the immunosuppressive "blanket" that was keeping the vasculitis quiet.
4. Anatomy and Function — Vessels Involved
Understanding which vessels EGPA targets explains the clinical features:
EGPA affects predominantly small vessels but can extend to medium-sized arteries:
- Capillaries (e.g., alveolar capillaries → pulmonary infiltrates; glomerular capillaries → GN)
- Venules (e.g., dermal venules → palpable purpura)
- Arterioles (e.g., vasa nervorum → mononeuritis multiplex)
- Small and medium arteries (e.g., coronary arteries → myocarditis/coronary vasculitis; mesenteric arteries → abdominal pain)
The vasa nervorum are the tiny blood vessels that supply peripheral nerves. When these are inflamed (vasculitis of the vasa nervorum), the nerve loses its blood supply → nerve infarction → mononeuritis multiplex. This is why peripheral neuropathy is so common in EGPA (up to 75%).
- Upper airway: nasal mucosa (allergic rhinitis, nasal polyps)
- Lower airway: bronchial smooth muscle and mucosa (asthma), alveolar parenchyma (eosinophilic pneumonia)
- Pulmonary vasculature: small vessel vasculitis → pulmonary infiltrates, rarely diffuse alveolar haemorrhage
5. Etiology and Pathophysiology
The exact cause of EGPA remains unknown (idiopathic). It is considered an autoimmune condition arising in a Th2-predominant immunological milieu in genetically susceptible individuals.
Proposed contributing factors:
- Genetic susceptibility: HLA-DRB104, HLA-DRB107; IL-10 promoter polymorphisms
- Environmental triggers: Allergens, infections, drugs (as above)
- Immune dysregulation: Exaggerated Th2 response → IL-4, IL-5, IL-13 → eosinophil activation and survival
- ANCA production (in ~40–50%): Anti-MPO antibodies → neutrophil and endothelial activation
5.2 Pathophysiology — Step by Step
EGPA pathophysiology can be conceptualised as two parallel but overlapping pathogenic arms:
Genetic predisposition + Environmental triggers
↓
Th2 immune polarisation
↓
↑ IL-4, IL-5, IL-13 production
↓
IL-5 → Eosinophil differentiation, activation, and survival
IL-4/IL-13 → IgE class switching (↑ total IgE)
↓
Eosinophil tissue infiltration
↓
Eosinophil degranulation → release of:
• Major basic protein (MBP)
• Eosinophil cationic protein (ECP)
• Eosinophil peroxidase (EPO)
• Eosinophil-derived neurotoxin (EDN)
↓
Direct tissue damage → granuloma formation
↓
End-organ damage (heart, lungs, GI tract, skin)Why granulomas form: When eosinophils and macrophages encounter persistent antigen/damaged tissue they cannot clear, macrophages transform into epithelioid cells and giant cells, walling off the insult → necrotizing granulomatous inflammation. The eosinophilic infiltrate gives the granulomas their characteristic "eosinophil-rich" appearance on histology.
Loss of immune tolerance → production of anti-MPO (p-ANCA) antibodies
↓
Priming of neutrophils by TNF-α / IL-1 (e.g., from infection)
↓
ANCA binds to MPO on primed neutrophil surface
↓
Neutrophil activation → degranulation, ROS production
↓
Neutrophils adhere to endothelium → endothelial injury
↓
Necrotizing small vessel vasculitis
↓
Pauci-immune glomerulonephritis, purpura, nerve infarctionANCA-positive EGPA patients have increased risk of renal involvement, peripheral neuropathy and biopsy-proven vasculitis [2]. ANCA-negative EGPA patients have higher eosinophilic count and tissue infiltration and increased risk of heart disease and parenchymal lung disease [2].
Two Phenotypes of EGPA — Exam Favourite
This dual-phenotype model is a very common exam question. ANCA-positive = more "vasculitic" (kidneys, nerves, purpura). ANCA-negative = more "eosinophilic" (heart, lungs). Both share asthma and eosinophilia as a common thread.
The disease classically evolves through three sequential phases, reflecting progressive immune dysregulation [1][2]:
| Phase | What Happens | Pathophysiology | Typical Duration |
|---|---|---|---|
| Phase 1: Prodromal (Allergic) | Asthma, allergic rhinitis, nasal polyposis, sinusitis | Th2 immune polarisation → IgE-mediated hypersensitivity; eosinophil recruitment to airways | Years to decades before vasculitis |
| Phase 2: Eosinophilic | Peripheral eosinophilia, eosinophilic tissue infiltration (pneumonia, gastroenteritis, myocarditis) | Marked eosinophil production (IL-5 driven) → tissue infiltration and degranulation → direct cytotoxic damage by eosinophil granule proteins | Months to years |
| Phase 3: Vasculitic | Systemic necrotizing vasculitis (mononeuritis multiplex, purpura, glomerulonephritis, cardiac vasculitis) | ANCA-mediated (in ANCA+ patients) and eosinophil-mediated endothelial injury → necrotizing vasculitis and granuloma formation | The "full-blown" disease |
Three phases: (1) Asthma and atopic allergies e.g. allergic rhinitis; (2) Eosinophilic infiltrative disease: pneumonia, esophagitis, GE; (3) Vasculitis: e.g. myocarditis [1]
Phase Overlap
Although classically described as sequential, in reality these phases frequently overlap. A patient may present with features of all three phases simultaneously. Don't be tricked into thinking they must occur one after another in a neat timeline.
Histology shows eosinophilic necrotizing granuloma [3][6]:
- Eosinophil-rich infiltrates surrounding blood vessel walls
- Fibrinoid necrosis of vessel walls (the "necrotizing" part)
- Granulomas with central necrotic core (sometimes with eosinophilic debris — "allergic granulomas"), surrounded by epithelioid histiocytes and multinucleated giant cells
- Pauci-immune (minimal/absent immunoglobulin and complement deposition on immunofluorescence) — this distinguishes AAV from immune-complex vasculitides
6. Classification
EGPA is classified under:
- Primary vasculitis → Small vessel vasculitis → ANCA-associated vasculitis (AAV)
Classification of primary vasculitis [6][7]:
| Vessel Size | Disease |
|---|---|
| Large vessel | Giant cell arteritis, Takayasu arteritis |
| Medium/small vessel | Polyarteritis nodosa (PAN) (Viral hepatitis B related) [7]; Kawasaki disease |
| Small vessel — ANCA-related | Granulomatosis with polyangiitis (Wegener's granulomatosis) (ANCA related); Eosinophilic granulomatosis with polyangiitis (Churg Strauss syndrome) (ANCA related); Microscopic polyangiitis (ANCA related) [7] |
| Small vessel — Immune complex | IgA vasculitis (Henoch-Schönlein purpura); Hypocomplementaemic urticarial vasculitis (anti-C1q vasculitis) [7] |
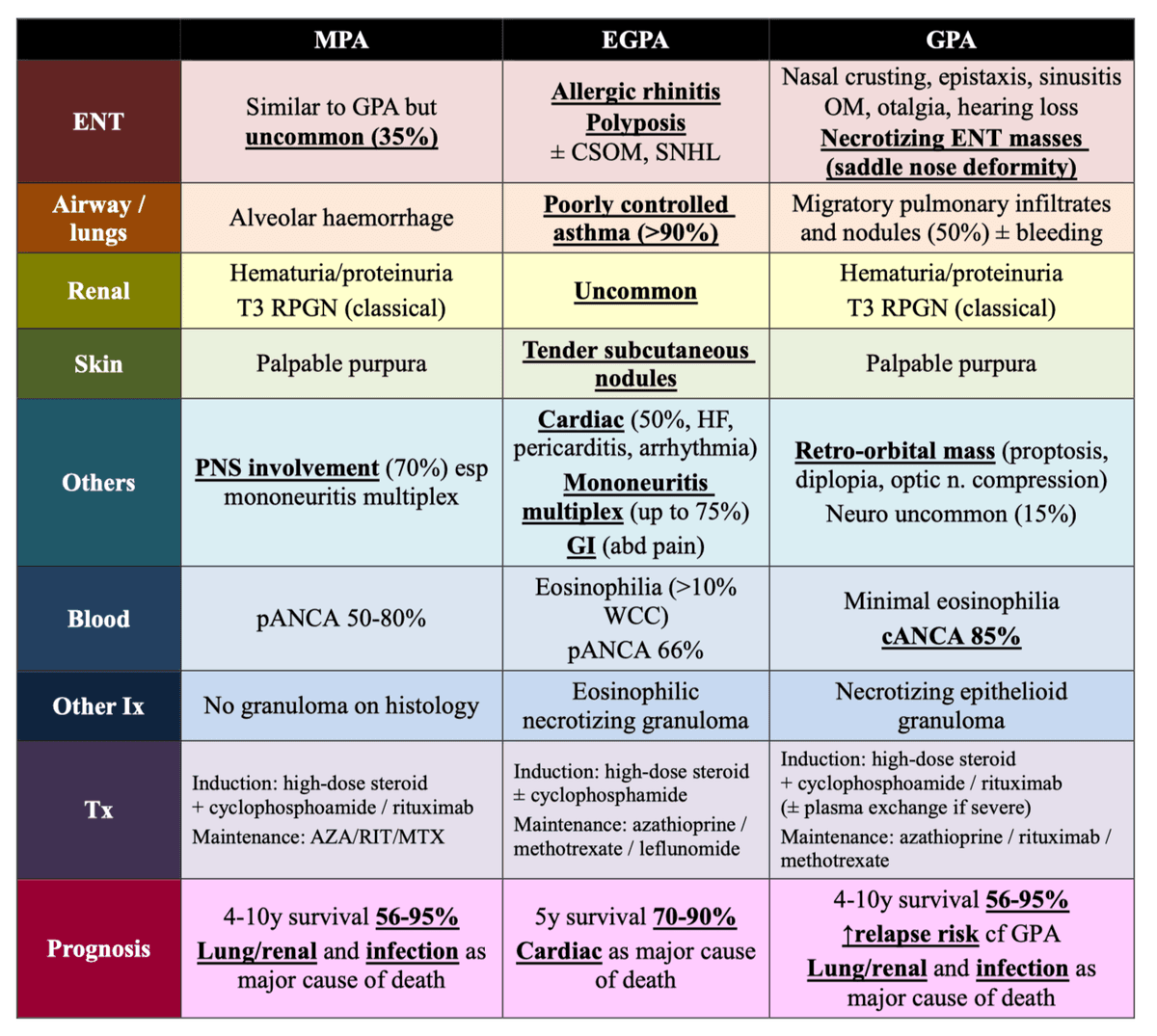
This table is extremely high yield for HKUMed exams as it appears in multiple senior notes and lecture slides [1][2][3][6]:
| Feature | MPA | EGPA (Churg-Strauss) | GPA (Wegener's) |
|---|---|---|---|
| Granuloma | −ve | +ve | +ve |
| Renal involvement | 90% | 45% | 80% |
| Pulmonary involvement | 50% | 70% (+ Asthma) | 90% (+ ENT) |
| Asthma | −ve | +ve | −ve |
| ANCA +ve rate | 70% | 40–50% | 90% |
| ANCA type | Anti-MPO (p-ANCA) | Anti-MPO (p-ANCA) | Anti-PR3 (c-ANCA) |
| ENT | Similar to GPA but uncommon | Rhinitis, polyposis [6] | Nasal crusting, epistaxis, sinusitis, saddle nose deformity, necrotizing lesions [6] |
| Eosinophilia | Minimal | > 10% of peripheral white cells [6] | Minimally elevated [6] |
| Histology | No granuloma; necrotizing vasculitis | Eosinophilic necrotizing granuloma [6] | Necrotizing epithelioid granuloma [6] |
| Major cause of death | Pulmonary and renal | Cardiac [6] | Pulmonary and renal [6] |
As discussed in pathophysiology, EGPA can be subdivided into two clinical phenotypes based on ANCA status:
| Feature | ANCA-positive (~40–50%) | ANCA-negative (~50–60%) |
|---|---|---|
| Dominant mechanism | Vasculitic (neutrophil-mediated) | Eosinophilic tissue infiltration |
| Renal involvement | More common (pauci-immune GN) | Less common |
| Peripheral neuropathy | More common | Less common |
| Purpura | More common | Less common |
| Cardiac involvement | Less common | More common (major cause of death) |
| Lung parenchymal disease | Less common | More common (eosinophilic pneumonia) |
| Eosinophil count | Lower | Higher |
| Biopsy-proven vasculitis | More common | Less common |
7. Clinical Features
Clinical features of EGPA should be understood through the lens of the three phases, recognising that overlap is the norm.
7.2 Symptoms
| Symptom | Pathophysiological Basis |
|---|---|
| Wheezing and dyspnoea (asthma) | Th2-mediated bronchial hyperresponsiveness → bronchospasm + mucus hypersecretion + airway wall remodelling. Poorly controlled asthma (> 90%) is the hallmark [3]. Often late-onset asthma (adult onset) that is difficult to control and progressively worsens |
| Nasal congestion/rhinorrhoea | Allergic rhinitis [1][6] — IgE-mediated mast cell degranulation in nasal mucosa → histamine release → mucosal oedema, rhinorrhoea |
| Nasal obstruction / anosmia | Nasal polyposis [3][6] — chronic eosinophilic inflammation of sinonasal mucosa → polypoid mucosal swelling |
| Facial pain/pressure | Chronic sinusitis — eosinophilic infiltration of sinus mucosa + obstruction from polyps |
| Symptom | Pathophysiological Basis |
|---|---|
| Cough, dyspnoea, fever (eosinophilic pneumonia) | Eosinophil infiltration of alveolar parenchyma → eosinophilic pneumonia with migratory pulmonary infiltrates |
| Dysphagia, abdominal pain, diarrhoea (eosinophilic GI disease) | Eosinophilic infiltrative disease: pneumonia, esophagitis, GE [1] — eosinophils infiltrate the oesophageal, gastric, or intestinal wall → mucosal inflammation, dysmotility, and rarely perforation |
| Chest pain/dyspnoea (cardiac) | Eosinophilic myocarditis — eosinophilic degranulation products (MBP, ECP) are directly toxic to cardiomyocytes → myocardial necrosis and fibrosis |
| Constitutional symptoms | Fever, malaise, weight loss, night sweats — driven by systemic cytokine release (IL-1, IL-6, TNF-α) from widespread inflammation |
| Symptom | Pathophysiological Basis |
|---|---|
| Numbness, tingling, weakness in specific nerve distributions (mononeuritis multiplex) | Vasculitis of the vasa nervorum → nerve ischaemia/infarction. Mononeuritis multiplex (up to 75%) [3]. Often presents as acute foot drop or wrist drop. Can be asymmetric and involve multiple non-contiguous nerves |
| Skin lesions (purpura, nodules) | Vasculitis of dermal venules → extravasation of red blood cells → palpable purpura. Eosinophilic granulomatous inflammation of subcutaneous tissue → tender subcutaneous nodules [3] |
| Haematuria, reduced urine output (renal) | Pauci-immune focal segmental necrotizing and crescentic glomerulonephritis (Type III RPGN) → haematuria, proteinuria, rising creatinine. Less common in EGPA (45%) than GPA/MPA |
| Chest pain, palpitations, syncope, dyspnoea (cardiac) | Cardiac involvement (50%) includes HF, pericarditis, arrhythmia [3]. Eosinophilic myocarditis → restrictive cardiomyopathy → heart failure. Coronary arteritis → ischaemia. Pericardial effusion → pericarditis |
| Abdominal pain (GI vasculitis) | Mesenteric vasculitis → bowel ischaemia → crampy abdominal pain, GI bleeding, rarely perforation. GI (abdominal pain) [3] |
| Arthralgia/myalgia | Synovial and muscular vessel vasculitis → inflammation. Non-deforming polyarthralgia is common |
| Haemoptysis (rare in EGPA) | Pulmonary capillaritis → diffuse alveolar haemorrhage (more common in MPA/GPA but can occur) |
7.3 Signs
| Sign | Pathophysiological Basis |
|---|---|
| Fever | Systemic inflammatory response; cytokine-mediated (IL-1, IL-6) |
| Weight loss / cachexia | Catabolic state from chronic systemic inflammation |
| Sign | Pathophysiological Basis |
|---|---|
| Bilateral polyphonic expiratory wheeze | Bronchospasm from asthma (Th2-driven airway hyperresponsiveness) |
| Prolonged expiratory phase | Air trapping from small airway obstruction |
| Nasal polyps on anterior rhinoscopy | Chronic eosinophilic inflammation of nasal mucosa → polypoid mucosal hypertrophy |
| Bibasal crackles (if eosinophilic pneumonia / alveolar haemorrhage) | Alveolar consolidation or fluid from eosinophilic infiltration or haemorrhage |
| Sign | Pathophysiological Basis |
|---|---|
| Tachycardia / arrhythmia | Eosinophilic myocarditis disrupts conduction system; pericarditis can cause sinus tachycardia |
| Elevated JVP, peripheral oedema, hepatomegaly (right heart failure signs) | Restrictive cardiomyopathy from myocardial fibrosis (endstage of eosinophilic myocarditis) → impaired diastolic filling → backward failure |
| S3 gallop | Dilated or failing ventricle with volume overload |
| Pericardial friction rub | Pericarditis — eosinophilic pericardial inflammation |
| Muffled heart sounds (if pericardial effusion) | Fluid between visceral and parietal pericardium dampens sound transmission |
| Sign | Pathophysiological Basis |
|---|---|
| Palpable purpura (especially lower limbs) | Leukocytoclastic vasculitis of dermal post-capillary venules → extravasation of RBCs into dermis. "Palpable" because inflammatory infiltrate elevates the lesions above skin surface |
| Tender subcutaneous nodules [3] | Eosinophilic granulomatous inflammation in subcutaneous tissue — similar pathology to erythema nodosum but with eosinophilic rather than neutrophilic predominance |
| Livedo reticularis | Net-like purplish discolouration from vasospasm or vasculitis of dermal arterioles → mottled blood flow pattern |
| Urticarial rash | IgE-mediated mast cell degranulation in skin → histamine → vasodilation and oedema |
| Sign | Pathophysiological Basis |
|---|---|
| Foot drop (common peroneal nerve palsy) | Vasculitis of vasa nervorum of common peroneal nerve → nerve infarction → loss of ankle dorsiflexion and eversion |
| Wrist drop (radial nerve palsy) | Same mechanism affecting radial nerve |
| Asymmetric sensory loss in named nerve territories | Mononeuritis multiplex: multiple discrete nerve infarctions from vasculitis of their vasa nervorum |
| Reduced or absent reflexes (in affected territories) | Lower motor neurone pattern from peripheral nerve damage |
| Cranial nerve palsies (rare) | Vasculitis of cranial nerve vasa nervorum |
| Sign | Pathophysiological Basis |
|---|---|
| Hypertension | Renal vasculitis → activation of RAAS from renal ischaemia |
| Peripheral oedema (if nephrotic-range proteinuria or advanced CKD) | Reduced oncotic pressure (hypoalbuminaemia) or fluid retention from declining GFR |
| Sign | Pathophysiological Basis |
|---|---|
| Episcleritis / Scleritis | Vasculitis of episcleral/scleral vessels → painful red eye |
| Peripheral ulcerative keratitis (PUK) | ANCA vasculitides (GPA, EGPA) are associated with PUK [8] — immune-complex deposition and complement activation at corneal periphery → MMP-mediated corneal breakdown |
| Organ System | Frequency | Key Features | Dominant Pathway |
|---|---|---|---|
| Respiratory | > 90% | Asthma (virtually always), allergic rhinitis, nasal polyps, sinusitis, eosinophilic pneumonia | Th2/Eosinophilic |
| Neurological | ~70–75% | Mononeuritis multiplex (foot drop, wrist drop), polyneuropathy, rarely CNS vasculitis | Vasculitic (vasa nervorum) |
| Cardiac | ~50% | Myocarditis, pericarditis, heart failure, arrhythmias, coronary arteritis | Eosinophilic infiltration (ANCA-negative phenotype dominant) |
| Skin | ~50–70% | Palpable purpura, subcutaneous nodules, urticaria, livedo reticularis | Both |
| GI | ~30–50% | Eosinophilic gastroenteritis, mesenteric vasculitis, abdominal pain, diarrhoea | Both |
| Renal | ~45% | Pauci-immune GN (Type III RPGN), haematuria, proteinuria | Vasculitic (ANCA-positive phenotype) |
| MSK | ~50% | Arthralgia, myalgia (non-deforming) | Inflammatory |
| Constitutional | Common | Fever, weight loss, fatigue | Cytokine-mediated |
10. Important Clinical Pearls for HKUMed Exams
Both present with asthma + eosinophilia + pulmonary infiltrates, but:
- ABPA: caused by hypersensitivity to Aspergillus fumigatus; ↑↑ total IgE (> 1000 IU/mL); positive Aspergillus skin test; central bronchiectasis; NO vasculitis or multi-organ involvement
- EGPA: systemic vasculitis; ANCA may be positive; multi-organ involvement (heart, nerves, kidneys, GI); granulomatous inflammation on biopsy
Leukotriene receptor antagonists (montelukast, zafirlukast) have steroid-sparing effect and can unmask previously undiagnosed Churg-Strauss Syndrome [5]. This is because:
- Patient has "difficult asthma" → actually prodromal EGPA
- They are on inhaled or oral corticosteroids which suppress the vasculitic/eosinophilic components
- Clinician adds LTRA and tapers steroids (since asthma seems controlled with LTRA)
- Steroid dose falls below the threshold needed to suppress the underlying vasculitis
- Systemic manifestations (eosinophilia, neuropathy, cardiac disease) emerge
Cardiac involvement is a common cause of morbidity and mortality [2]. Major cause of death in EGPA = cardiac [3][6]. Unlike GPA and MPA where renal and pulmonary disease dominate mortality, EGPA patients die of eosinophilic myocarditis, cardiac fibrosis, and arrhythmias.
High Yield Summary
Definition: EGPA (Churg-Strauss) = ANCA-associated necrotizing small-to-medium vessel vasculitis with eosinophil-rich granulomatous inflammation, asthma, and eosinophilia.
Epidemiology: Rare (0.5–6.8/million/year); age 20–40; slight male predominance.
Three Phases: (1) Allergic/Prodromal — asthma, allergic rhinitis, nasal polyps; (2) Eosinophilic — tissue infiltration (pneumonia, GI, myocarditis); (3) Vasculitic — mononeuritis multiplex, purpura, GN.
Key ANCA: p-ANCA (anti-MPO) positive in 40–50%. ANCA+ = more vasculitic (renal, nerves). ANCA− = more eosinophilic (cardiac, lung parenchyma).
Comparison Table (GPA vs EGPA vs MPA): EGPA is the only AAV with asthma and marked eosinophilia (> 10% WCC). Cardiac death is the major cause of mortality in EGPA (vs pulmonary/renal in GPA/MPA).
Histology: Eosinophilic necrotizing granuloma; pauci-immune on IF.
LTRA alert: Montelukast unmasks EGPA when steroids are tapered.
Cardinal clinical features: Asthma (> 90%), mononeuritis multiplex (75%), cardiac involvement (50%), eosinophilia (> 10%), palpable purpura, subcutaneous nodules.
Active Recall - EGPA (Churg-Strauss Syndrome)
[1] Senior notes: Maksim Medicine Notes.pdf (Rheumatology, p.331) [2] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (Rheumatological Diseases — EGPA, pp.1767–1769) [3] Senior notes: Ryan Ho Rheumatology.pdf (Vasculitis section, pp.93–97) [4] Senior notes: Ryan Ho Respiratory.pdf (Pulmonary Eosinophilia and Vasculitides, pp.139–140) [5] Lecture slides: GC 040. Cough and wheezing_asthma and allergic lung diseases.pdf (p.40 — Leukotriene receptor antagonists) [6] Lecture slides: GC 053. Fingers turn white and blue.pdf (pp.79–80, p.94 — EGPA vs GPA comparison table; Classification of primary vasculitis) [7] Senior notes: Ryan Ho Urogenital.pdf (ANCA-associated vasculitis, p.68) [8] Senior notes: Ryan Ho Opthalmology.pdf (PUK and rheumatological disease, p.131)
Differential Diagnosis of EGPA (Churg-Strauss Syndrome)
The differential diagnosis of EGPA is challenging because its features — asthma, eosinophilia, pulmonary infiltrates, multisystem inflammation — overlap with a wide range of conditions. The key to organising the DDx is to think about which presenting feature dominates and then systematically work through conditions that mimic that feature. Below we organise DDx by the major clinical "entry points" through which a patient with suspected EGPA might present.
A patient with suspected EGPA may present through one (or more) of these "doors":
- Difficult / late-onset asthma with eosinophilia — the prodromal phase
- Peripheral blood eosinophilia — the haematological finding
- Pulmonary infiltrates with eosinophilia — pulmonary eosinophilia
- Multi-organ vasculitis — the vasculitic phase (skin, nerves, kidneys, heart)
- Rapidly progressive glomerulonephritis (RPGN) — the renal presentation
Each door has its own DDx list. True EGPA sits at the intersection of multiple lists.
2. Differential Diagnosis by Presenting Feature
This is the most treacherous differential because EGPA masquerades as difficult-to-control asthma for years before the vasculitic phase emerges.
Churg-Strauss syndrome is an ANCA-related vasculitis. It may present as asthma in its early stages but may progress to widespread eosinophilic vasculitic damage to various organs. It is commonly mis-diagnosed as asthma and coincidentally can be treated by ICS. [5]
| Differential | Key Distinguishing Features | Why It's NOT EGPA |
|---|---|---|
| Atopic asthma | Childhood onset, family/personal history of atopy (eczema, allergic rhinitis, allergic conjunctivitis), well-controlled with standard therapy, eosinophilia mild (< 1.0 × 10⁹/L) | No vasculitic features, no multi-organ disease, no granulomas. Eosinophilia is modest and resolves with ICS |
| Allergic bronchopulmonary aspergillosis (ABPA) [9] | Asthma + ↑↑ Total IgE (typically > 1000 IU/mL) + positive Aspergillus skin test / ↑ IgE against Aspergillus fumigatus + central bronchiectasis + mucoid impaction | ABPA is an allergic response to Aspergillus fumigatus colonising the airways. There is NO vasculitis, no mononeuritis multiplex, no cardiac involvement. ANCA is negative. Responds to steroids ± itraconazole |
| Aspirin/NSAIDs-exacerbated respiratory disease (AERD) [9] | Samter's triad = Asthma + Aspirin sensitivity + Nasal polyps. COX-1 inhibition shunts arachidonic acid to the lipo-oxygenase pathway → excess leukotrienes | No systemic vasculitis, no peripheral eosinophilia > 10%, no cardiac or renal disease. History of aspirin/NSAID-triggered bronchospasm is the clue |
| Severe eosinophilic asthma (T2-high) | Eosinophilic airway inflammation with sputum and blood eosinophilia, responds to anti-IL-5 biologics (mepolizumab) | No extra-pulmonary organ involvement. Important: one must actively exclude EGPA before diagnosing "severe eosinophilic asthma" by looking for vasculitic features |
Exam Trap — LTRA Unmasking
When such a patient switches from steroids to LTRA, systemic manifestations will resurface [5]. If an exam stem describes an asthmatic patient developing mononeuritis multiplex or cardiac symptoms after starting montelukast and reducing steroids, think EGPA being unmasked, not a new disease.
The classic mnemonic for eosinophilia is NAACP [10][11]:
| Letter | Category | Key Differentials | How to Distinguish from EGPA |
|---|---|---|---|
| N | Neoplasm | Hypereosinophilic syndrome (HES), acute/chronic eosinophilic leukaemia, lymphoma, solid tumours (esp adenoCA of GI, lung) [10] | HES: sustained eosinophilia > 1.5 × 10⁹/L for > 6 months with end-organ damage but WITHOUT vasculitis or granulomas. May have FIP1L1-PDGFRA fusion (responds to imatinib). No asthma history |
| A | Addison's disease | Classical teaching but usually only if acutely ill [10] | Adrenal insufficiency causes mild eosinophilia via loss of cortisol's eosinophil-suppressive effect. Look for hypotension, hyperpigmentation, ↑K⁺, ↓Na⁺, ↓glucose |
| A | Allergy | Allergic rhinitis, asthma, eczema, drug allergy, ABPA [10] | Allergic eosinophilia is usually modest (< 1.5 × 10⁹/L). No vasculitic features |
| C | Collagen vascular disease | EGPA (Churg-Strauss), other CTDs [10] | EGPA itself sits here. Other CTDs (SLE, RA, dermatomyositis) can cause mild eosinophilia but don't produce the classic asthma + eosinophilia + granulomatous vasculitis triad |
| P | Parasitic disease | Especially helminths (Strongyloides, Schistosoma) [10] | Helminth infections can cause dramatic eosinophilia and pulmonary infiltrates (Löffler syndrome). Travel history, stool O&P, specific serology are key. No vasculitis on biopsy. Hong Kong relevance: consider Strongyloides stercoralis in immigrants from Southeast Asia |
Hypereosinophilic Syndrome vs EGPA — Critical Distinction
HES = eosinophilia > 1.5 × 10⁹/L for > 6 months + end-organ damage (cardiac fibrosis, CNS, skin) without vasculitis, granulomas, or pre-existing asthma. EGPA = eosinophilia + asthma + granulomatous vasculitis. Both can cause eosinophilic myocarditis, which is why the distinction matters — treatment differs (imatinib may work in HES with PDGFRA fusion; EGPA needs immunosuppression).
This is a crucial DDx domain because EGPA causes migratory pulmonary infiltrates [3].
| Differential | Features | Why It's NOT EGPA |
|---|---|---|
| Acute eosinophilic pneumonia (AEP) | Acute febrile illness ( < 7 days), respiratory failure, bilateral diffuse GGO on HRCT, BAL > 25% eosinophils. Often young male, new-onset smoker [4] | Acute and self-limiting. No asthma history, no vasculitis, no multi-organ disease. Excellent response to steroids with no relapse |
| Chronic eosinophilic pneumonia (CEP) | Insidious onset, middle-aged female, bilateral peripheral upper-lobe opacities ("photographic negative of pulmonary oedema"), BAL ≥ 25% eosinophils, ↑ ESR, ↑ IgE [4] | No vasculitis — purely a parenchymal disease. No mononeuritis multiplex, no cardiac involvement, ANCA negative. Responds to prednisolone but tends to relapse when steroids are tapered |
| Löffler syndrome | Transpulmonary passage of helminth larvae with transient CXR opacities and eosinophilia [4]. Caused by Ascaris lumbricoides, Strongyloides, hookworm | Self-limiting (2–4 weeks). Travel history and stool O&P are diagnostic. No vasculitis |
| Tropical pulmonary eosinophilia | Immune response to microfilariae of Wuchereria bancrofti [4]. Mixed restrictive-obstructive pattern, diffuse opacities, cough, SOB, wheeze | Endemic area travel history; responds to diethylcarbamazine. No vasculitis |
| Drug-induced pulmonary eosinophilia | Drugs: NSAIDs, nitrofurantoin, tryptophan, sulphonamides [4] | Temporal relationship with drug exposure. Resolves on drug withdrawal. No systemic vasculitis |
| ABPA | Central bronchiectasis, mucoid impaction, ↑↑ IgE, positive Aspergillus serology [9] | Fungal-driven. No vasculitis |
2D. Systemic Vasculitis — The Other Vasculitides
This is where the DDx becomes most exam-relevant. When a patient presents with multi-organ vasculitis, you must differentiate EGPA from the other ANCA-associated vasculitides and from other systemic vasculitides.
This comparison table from GC lecture slides is extremely high yield [6]:
Eosinophilic granulomatosis polyangiitis vs Granulomatosis polyangiitis [6]:
| Feature | EGPA | GPA | MPA |
|---|---|---|---|
| ENT | Rhinitis, polyposis | Necrotising lesions (saddle nose, sinusitis, nasal crusting, epistaxis) | Similar to GPA but uncommon (35%) |
| Allergy / bronchial asthma | Frequent | Similar to general population | Absent |
| Renal involvement | Uncommon (45%) | Common (80%) | Very common (90%) — T3 RPGN classical [3] |
| Eosinophilia | > 10% of peripheral white cells | Minimally elevated | Minimal |
| ANCA | pANCA (anti-MPO) 66% | cANCA (anti-PR3) 85% | pANCA (anti-MPO) 50–80% |
| Histology | Eosinophilic necrotising granuloma | Necrotising epithelioid granuloma | No granuloma |
| Major cause of death | Cardiac | Pulmonary and renal | Pulmonary and renal |
Key distinguishing logic:
- Asthma present? → EGPA (neither GPA nor MPA has asthma)
- ENT destruction (saddle nose)? → GPA (EGPA has allergic rhinitis/polyps, not destruction)
- Eosinophilia > 10%? → EGPA
- RPGN dominant? → MPA or GPA (less common in EGPA)
- c-ANCA (anti-PR3)? → GPA
- p-ANCA (anti-MPO)? → EGPA or MPA (distinguish by asthma and eosinophilia)
| Differential | Key Features | Why It's NOT EGPA |
|---|---|---|
| Polyarteritis nodosa (PAN) [1] | Necrotizing vasculitis of medium/small arteries, NOT associated with ANCA [1]. HBV-related. Livedo reticularis, digital ulcers/gangrene, mononeuritis multiplex, orchitis, MI, renal HT | PAN is ANCA-negative. No asthma, no eosinophilia, no pulmonary involvement (spares lungs — this is a classic distinguishing point). Orchitis is relatively specific for PAN. HBsAg may be positive |
| IgA vasculitis (Henoch-Schönlein purpura) | Vasculitis with IgA1-dominant immune deposits affecting small vessels [2]. Palpable purpura + abdominal pain + arthritis + GN. Mostly children | IgA deposition on IF (not pauci-immune). No asthma, no eosinophilia. Normal ANCA. Complement normal. Serum IgA may be elevated |
| SLE vasculitis | Multisystem autoimmune disease. Malar rash, photosensitivity, oral ulcers, arthritis, serositis, nephritis, cytopenias. ANA+, anti-dsDNA+ | Characteristic rash and arthritis suggest SLE [12]. SLE causes immune-complex GN (granular IF with ↓ C3/C4), not pauci-immune GN. No asthma, no marked eosinophilia |
| Cryoglobulinaemic vasculitis | Vasculitis with cryoglobulin immune deposits; skin, glomeruli, and peripheral nerves often involved [2]. HCV-associated (most common cause). Palpable purpura, arthralgia, GN, neuropathy | Cryocrit positive, ↓ C4 (classic), ↓ C3. HCV serology usually positive. Granular IF. No asthma, no eosinophilia |
| Sarcoidosis | Non-caseating granulomas, hilar lymphadenopathy, ↑ ACE, ↑ Ca²⁺, uveitis, erythema nodosum [2] | Sarcoidosis granulomas are NON-caseating and non-eosinophilic (unlike the eosinophilic necrotising granulomas of EGPA). No vasculitis. No asthma (though airway involvement can cause wheeze). ANCA negative |
When EGPA presents with RPGN, the DDx is structured by immunofluorescence (IF) pattern on renal biopsy [13][14][15]:
| IF Pattern | Type | Diseases | How to Distinguish |
|---|---|---|---|
| Negative / Pauci-immune | Type III | ANCA-associated vasculitis: GPA, EGPA, MPA [13][14] | Within this group, EGPA is distinguished by asthma + eosinophilia + p-ANCA (anti-MPO). GPA has ENT destruction + c-ANCA. MPA has no granulomas |
| Linear | Type I | Anti-GBM disease / Goodpasture syndrome [13][14] | Linear IgG deposition along GBM. Anti-GBM antibody positive. Goodpasture = GN + pulmonary haemorrhage (haemoptysis). No asthma, no eosinophilia |
| Granular | Type II | Immune complex GN: SLE, PSGN, IgAN, MPGN, cryoglobulinaemia, IE [13][14] | Granular deposits of Ig/C3. ↓ C3/C4 in lupus, PSGN, cryoglobulinaemia, MPGN [12]. Normal C3/C4 in IgAN/HSP [12]. No asthma, no eosinophilia |
Serum complement level: important in helping narrow the differential diagnosis. ↓ C3/4 generally indicates IC-mediated GN. Normal C3/4 generally indicates non-IC-mediated GN [12]
When EGPA presents with foot drop or wrist drop from mononeuritis multiplex, the DDx includes [16]:
Causes of mononeuritis multiplex: Wegener's granulomatosis, Amyloidosis, Rheumatoid arthritis, Diabetes mellitus, SLE, Polyarteritis nodosa, Leprosy, Carcinomatosis and Churg-Strauss syndrome [16]
The mnemonic is essentially a list of vasculitic and infiltrative neuropathies:
- DM: most common cause overall — diabetic amyotrophy from vasa nervorum disease (atherosclerotic, not vasculitic)
- PAN: medium vessel vasculitis → vasa nervorum infarction (ANCA-negative, no asthma)
- GPA / MPA: other AAVs
- SLE / RA: secondary vasculitis
- Leprosy: consider in Hong Kong patients from endemic areas (Philippines, mainland China)
- Amyloidosis: amyloid infiltration of vasa nervorum
- Carcinomatous neuropathy: paraneoplastic
The key distinguishing feature for EGPA: asthma + eosinophilia + p-ANCA + vasculitic neuropathy is a combination that no other condition produces.
| DDx Category | Condition | Asthma | Eosinophilia | Vasculitis | ANCA | Granuloma | Cardiac | Key Distinguishing Feature |
|---|---|---|---|---|---|---|---|---|
| AAV | GPA | − | Mild | + | c-ANCA (PR3) | + | Rare | ENT destruction, saddle nose |
| AAV | MPA | − | Mild | + | p-ANCA (MPO) | − | Rare | RPGN dominant, no granuloma |
| Medium vessel | PAN | − | − | + | − | − | MI | Orchitis, spares lungs, HBV |
| IC vasculitis | IgA vasculitis | − | − | + | − | − | − | Children, IgA on IF |
| IC vasculitis | Cryoglobulinaemic | − | − | + | − | − | − | ↓ C4, HCV, cryocrit+ |
| Secondary | SLE | − | − | ± | − | − | Serositis | ANA/dsDNA+, ↓ C3/C4 |
| Pulm eosinophilia | AEP | − | + | − | − | − | − | Acute (< 7d), self-limiting |
| Pulm eosinophilia | CEP | − | + | − | − | − | − | Peripheral upper-lobe infiltrates |
| Pulm eosinophilia | Löffler | − | + | − | − | − | − | Helminth larvae, transient |
| Allergic | ABPA | + | + | − | − | − | − | Aspergillus serology, ↑↑ IgE |
| Allergic | AERD | + | Mild | − | − | − | − | Aspirin-triggered, nasal polyps |
| Haematological | HES | − | ++ | − | − | − | + | No asthma, no vasculitis, FIP1L1-PDGFRA |
| Granulomatous | Sarcoidosis | − | − | − | − | + (non-caseating) | + | ↑ ACE, ↑ Ca²⁺, hilar LN |
| Anti-GBM | Goodpasture | − | − | + | − | − | − | Linear IF, anti-GBM Ab, haemoptysis |
The practical clinical approach when confronted with a suspected EGPA case is:
- Confirm asthma: Nearly universal in EGPA. If absent, think GPA, MPA, PAN, HES instead
- Check eosinophil count: EGPA typically > 1.5 × 10⁹/L (often > 10% of WCC). If eosinophilia is absent or mild, consider GPA/MPA
- Check ANCA: p-ANCA (anti-MPO) in ~40–50%. If c-ANCA (anti-PR3), think GPA. If ANCA negative with eosinophilia, consider ANCA-negative EGPA vs HES
- Look for extra-pulmonary organ involvement: Mononeuritis multiplex, cardiac disease, purpura, GN → supports vasculitic phase of EGPA
- Check complement levels: ↓ C3/C4 → immune complex-mediated GN (lupus, PSGN, cryoglobulinaemia); normal C3/C4 → pauci-immune (ANCA-associated) or anti-GBM [12]
- Exclude parasites: Stool O&P, Strongyloides serology — essential in Hong Kong given Southeast Asian migration
- Exclude ABPA: Aspergillus skin test, specific IgE, total IgE
- Biopsy: Eosinophilic necrotising granuloma + pauci-immune IF → confirms EGPA
- GPA vs EGPA: GPA destroys the upper airway (necrotising granulomas erode cartilage → saddle nose); EGPA merely inflames it allergically (rhinitis, polyps). GPA has no asthma. GPA is c-ANCA; EGPA is p-ANCA
- MPA vs EGPA: MPA has no granulomas at all on histology. MPA has no asthma. MPA is dominated by RPGN and pulmonary haemorrhage
- PAN vs EGPA: PAN is ANCA-negative and spares the lungs (no pulmonary involvement). PAN has orchitis (testicular artery vasculitis), which EGPA essentially never does. PAN affects medium-sized arteries
- HES vs EGPA: HES lacks asthma and vasculitis. HES is a primary bone marrow disorder of eosinophil overproduction (often clonal with FIP1L1-PDGFRA). Both can cause eosinophilic myocarditis, making the distinction critical for treatment
- CEP vs EGPA: CEP is a purely pulmonary disease — no vasculitis, no multi-organ involvement. It responds beautifully to steroids but relapses
- Sarcoidosis vs EGPA: Sarcoidosis granulomas are non-caseating and non-eosinophilic (composed of epithelioid histiocytes and Langhans giant cells without eosinophils). Sarcoidosis causes ↑ ACE and ↑ Ca²⁺. No asthma, no eosinophilia, no ANCA
High Yield Summary — DDx of EGPA
-
The unique triad of EGPA = Asthma + Eosinophilia (> 10% WCC) + Granulomatous vasculitis. No other condition produces all three together.
-
Among AAVs: EGPA is the only one with asthma and marked eosinophilia. GPA has ENT destruction + c-ANCA. MPA has no granulomas.
-
Key mimics to exclude: ABPA (Aspergillus serology, ↑↑ IgE, no vasculitis), HES (no asthma, no vasculitis, may have FIP1L1-PDGFRA), CEP (no vasculitis), parasitic eosinophilia (travel history, stool O&P).
-
For RPGN DDx: Use IF pattern — pauci-immune (Type III) = AAV; linear (Type I) = anti-GBM; granular (Type II) = immune complex. Complement levels help: ↓ C3/C4 = immune complex; normal = pauci-immune/anti-GBM.
-
LTRA unmasking: An asthmatic who develops systemic symptoms after steroid taper + LTRA initiation should be investigated for EGPA.
-
Cardiac cause of death in EGPA (vs pulmonary/renal in GPA/MPA) — this difference is frequently tested.
Active Recall - DDx of EGPA
References
[1] Senior notes: Maksim Medicine Notes.pdf (Rheumatology — PAN and ANCA-associated vasculitis, p.331) [2] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (Rheumatological Diseases — EGPA, GPA, MPA, small vessel vasculitis classification, pp.1764–1775) [3] Senior notes: Ryan Ho Rheumatology.pdf (Vasculitis — AAV comparison table, pp.93–97) [4] Senior notes: Ryan Ho Respiratory.pdf (Pulmonary Eosinophilia and Vasculitides, pp.139–140) [5] Senior notes: Ryan Ho Respiratory.pdf (LTRA and Churg-Strauss, footnote 73, p.106); Lecture slides: GC 040. Cough and wheezing_asthma and allergic lung diseases.pdf (p.40) [6] Lecture slides: GC 053. Fingers turn white and blue.pdf (pp.79–80, p.94 — EGPA vs GPA comparison table) [9] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (Asthma-associated syndromes — ABPA, AERD, EGPA, p.173) [10] Senior notes: Ryan Ho Haemtology.pdf (Eosinophilia — NAACP mnemonic, p.46) [11] Senior notes: Ryan Ho Fundamentals.pdf (Eosinophilia — NAACP, p.389) [12] Senior notes: Ryan Ho Fundamentals.pdf (Nephritic syndrome evaluation — complement levels, serology, pp.359–361) [13] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (RPGN classification by IF pattern, pp.413–421) [14] Senior notes: Ryan Ho Fundamentals.pdf (RPGN classification — Type I/II/III, p.361) [15] Senior notes: Ryan Ho Urogenital.pdf (ANCA-associated GN, p.68) [16] Senior notes: Ryan Ho Neurology.pdf (Mononeuritis multiplex causes, p.180)
Diagnostic Criteria, Diagnostic Algorithm and Investigations for EGPA
1. Diagnostic Criteria
There is no single universally agreed "gold-standard" diagnostic criteria set for EGPA. Two main frameworks exist: the classic 1990 ACR Classification Criteria and the newer 2022 ACR/EULAR Classification Criteria. Additionally, the Chapel Hill Consensus Conference (CHCC 2012) provides a pathological definition. Understanding the evolution of these criteria — and their limitations — is essential.
These were originally designed for classification (i.e., categorising a patient already known to have vasculitis), NOT for diagnosis in an unselected patient. However, they remain widely referenced in clinical practice and exams.
≥ 4 of 6 criteria → classifies as EGPA (sensitivity 85%, specificity 99.7%):
| # | Criterion | Rationale / Why This Criterion |
|---|---|---|
| 1 | Asthma (history of wheezing or diffuse high-pitched expiratory sounds) | Asthma is almost universal in EGPA — the Th2-driven prodromal phase. Distinguishes EGPA from GPA/MPA |
| 2 | Eosinophilia > 10% on differential WBC count | Reflects the marked IL-5-driven eosinophil production. Eosinophilia > 10% of peripheral white cells [6] |
| 3 | Mononeuropathy or polyneuropathy | Vasculitis of vasa nervorum → nerve infarction is one of the most characteristic vasculitic features |
| 4 | Non-fixed pulmonary infiltrates on CXR (migratory) | Eosinophilic pneumonia causes transient, migratory parenchymal opacities (unlike fixed consolidation from infection) |
| 5 | Paranasal sinus abnormality | Chronic sinusitis from eosinophilic mucosal inflammation. Distinguishes from "pure" asthma |
| 6 | Extravascular eosinophils on biopsy (including artery, arteriole, venule) | The histological hallmark — eosinophilic infiltration of tissue beyond the blood vessel wall |
ACR Criteria — Important Caveat
These criteria were designed to classify patients already known to have vasculitis, not to screen the general population. They should not be used as a screening tool in isolation. A patient may have genuine EGPA but not meet 4/6 criteria (e.g., early disease before biopsy). Conversely, a patient with HES + asthma could theoretically fulfil 4/6 without having true vasculitis.
These updated criteria use a weighted scoring system and were developed to improve discrimination from GPA, MPA, and other eosinophilic conditions. They should be applied only after a diagnosis of small- or medium-vessel vasculitis has been established (i.e., the patient is already confirmed to have vasculitis, and you are asking "which type?").
Entry criterion: Diagnosis of small/medium vessel vasculitis established
Step 1: Exclude mimics (especially HES, parasitic disease, drug reaction)
Step 2: Apply weighted scoring:
| Criterion | Points |
|---|---|
| Clinical criteria | |
| Obstructive airway disease (asthma or equivalents) | +3 |
| Nasal polyps | +3 |
| Mononeuritis multiplex | +1 |
| Laboratory criteria | |
| Eosinophil count ≥ 1 × 10⁹/L | +5 |
| Histopathological criteria | |
| Extravascular eosinophilic-predominant inflammation | +2 |
| Negative criterion (reduces score) | |
| Positive c-ANCA or anti-PR3 | −3 |
| Haematuria | −1 |
Score ≥ 6 → classified as EGPA
Key features of the 2022 system:
- Eosinophil count ≥ 1 × 10⁹/L carries the heaviest weight (+5) — reflecting that marked eosinophilia is the single most discriminating feature
- Positive c-ANCA / anti-PR3 is a negative criterion (−3) — because c-ANCA/PR3 overwhelmingly suggests GPA rather than EGPA. EGPA is associated with p-ANCA (anti-MPO) [1][2][6], not c-ANCA
- Haematuria subtracts points (−1) because prominent renal disease is more typical of GPA/MPA
Eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract and associated with asthma and eosinophilia. Necrotizing vasculitis predominantly affecting small to medium vessels. ANCA is more frequent when glomerulonephritis is present [2]
This is a pathological description, not a point-based criterion. It tells you what EGPA is at the tissue level.
In real life, the diagnosis is made by clinical pattern recognition + supportive investigations + biopsy when possible:
- History of late-onset/difficult-to-control asthma with allergic rhinitis/nasal polyps
- Peripheral blood eosinophilia > 1.5 × 10⁹/L (often > 10% WCC)
- Multi-organ involvement consistent with vasculitis (neuropathy, cardiac, skin, renal, GI)
- p-ANCA (anti-MPO) positive in ~40–50% [2][3]
- Biopsy of involved organ showing eosinophilic necrotizing granuloma and/or necrotizing vasculitis [2][17]
- Exclusion of mimics (parasites, HES, ABPA, drug reaction)
The following algorithm represents the systematic approach from clinical suspicion to confirmed diagnosis:
3. Investigation Modalities — Systematic Breakdown
| Investigation | Key Findings in EGPA | Interpretation / Why |
|---|---|---|
| CBC with differentials [1][2][17] | Eosinophilia — often > 1.5 × 10⁹/L, frequently > 10% of peripheral WCC [6]. May be > 5–10 × 10⁹/L in active disease. May also have normocytic normochromic anaemia (anaemia of chronic disease) | IL-5-driven eosinophil production. The degree of eosinophilia reflects disease activity (falls with steroid treatment). Anaemia reflects chronic inflammation (hepcidin-mediated iron sequestration) |
| ESR and CRP [1][17] | ↑ ESR and CRP [17] — both elevated, sometimes markedly | Non-specific acute-phase reactants reflecting systemic inflammation. CRP rises due to IL-6-driven hepatic synthesis |
| RFT (Urea, Creatinine, eGFR) [1][17] | May show ↑ creatinine if renal involvement (GN) | Monitors for renal vasculitis. Creatinine rises when GFR falls — a rapidly rising creatinine should prompt urgent consideration of RPGN |
| LFT | Usually normal; may be deranged if hepatic vasculitis (rare) or drug-related | Baseline before immunosuppressive therapy |
| Cardiac biomarkers (cTn, CK, NT-proBNP) [18] | ↑ cTn indicates myocardial injury (eosinophilic myocarditis). ↑ NT-proBNP in heart failure | Cardiac involvement is a common cause of morbidity and mortality [2]. Troponin rise in the context of eosinophilia + asthma should prompt immediate investigation for eosinophilic myocarditis |
| Total IgE | Often elevated (reflects Th2 polarisation), but not as dramatically as in ABPA (where > 1000 IU/mL is typical) | Helps distinguish from ABPA. In EGPA, IgE is elevated but rarely > 1000 IU/mL. In ABPA, ↑ Total IgE level (typically > 1000 IU/mL) [9] |
| Serum complement (C3, C4) | Normal in EGPA | Normal C3/C4 generally indicates non-IC-mediated GN [12]. This is because EGPA causes pauci-immune vasculitis (no immune complex consumption of complement). If C3/C4 are low, think lupus, PSGN, cryoglobulinaemia instead |
Complement Levels — A Pivotal Discriminator
Serum complement level: important in helping narrow the differential diagnosis [12]. In EGPA, complement is NORMAL because it is a pauci-immune vasculitis (ANCA-mediated, not immune-complex-mediated). Low complement should redirect you towards immune complex GN (SLE, PSGN, MPGN, cryoglobulinaemia).
| Investigation | Key Findings | Interpretation |
|---|---|---|
| ANCA and subtypes [1][17] | p-ANCA (anti-MPO) positive in ~40–50% of EGPA patients [2]. Perinuclear ANCA (P-ANCA) (Anti-MPO) in 60% of ANCA-positive patients and Cytoplasmic ANCA (C-ANCA) (Anti-PR3) in 10% of patients [17] | ANCA is tested by two methods: (1) Indirect immunofluorescence (IIF) on ethanol-fixed neutrophils → gives the p-ANCA or c-ANCA pattern; (2) ELISA → identifies the specific antigen (anti-MPO or anti-PR3). Both should be done. A positive p-ANCA/anti-MPO supports EGPA or MPA. A positive c-ANCA/anti-PR3 strongly suggests GPA |
| ANA, anti-dsDNA [12] | Should be negative in EGPA | Ordered to exclude SLE. If positive with low complement, redirect to lupus nephritis |
| Anti-GBM antibody [12] | Should be negative | Ordered to exclude anti-GBM disease / Goodpasture syndrome. If positive with haemoptysis + RPGN, consider anti-GBM disease |
ANCA and its subtypes for ANCA-vasculitis; ANA, anti-dsDNA for lupus nephritis; anti-GBM autoAb for anti-GBM disease [12]
ANCA — Suggestive but NOT Diagnostic
Serum ANCA: suggestive but not diagnostic (can have both false positives or false negatives) [15]. A negative ANCA does NOT exclude EGPA (50–60% are ANCA-negative). Conversely, ANCA can be positive in other conditions (infections, drugs, IBD). Always correlate with the clinical picture and biopsy.
| Finding | Interpretation |
|---|---|
| Proteinuria [17] | Glomerular damage → protein leak through damaged GBM |
| Haematuria [17] | Glomerular haematuria indicated by dysmorphic RBCs (RBCs damaged passing through inflamed glomeruli) |
| RBC casts [17] | Pathognomonic of glomerulonephritis — RBCs trapped in Tamm-Horsfall protein casts within tubules |
| Dysmorphic RBCs [17] | Glomerular origin (vs smooth RBCs from lower urinary tract bleeding) |
| Sterile pyuria | White cells in urine without infection → active GN |
Urinalysis: screen for glomerulonephritis. Look for proteinuria, haematuria, sediment with RBC casts and dysmorphic RBCs [17]
| Modality | Findings in EGPA | Interpretation / Why |
|---|---|---|
| CXR | Bilateral patchy, migratory, non-segmental pulmonary infiltrates. No cavitation (unlike GPA). Occasionally pleural effusion | Migratory pulmonary infiltrates and nodules (50%) [3]. The migratory nature reflects eosinophilic pneumonia — eosinophils infiltrate one area, resolve, then appear elsewhere. Lack of cavitation differentiates from GPA (where cavitary nodules are classic) |
| HRCT thorax | Ground-glass opacities (GGO), consolidation (often peripheral), air trapping on expiratory images (from asthma), septal thickening. Rarely nodules; almost never cavitation | More sensitive than CXR. GGO reflects active eosinophilic alveolitis. Air trapping reflects chronic asthma. Absence of cavitation is an important negative finding (would suggest GPA if present) |
| CT sinuses | Mucosal thickening, sinus opacification, nasal polyps | Documents chronic rhinosinusitis / polyposis — supports prodromal allergic phase |
| Echocardiography [18] | Regional/global LV dysfunction, ↓ LVEF, pericardial effusion, thickened myocardium (eosinophilic infiltration), valvular regurgitation, intracardiac thrombus | First-line cardiac assessment. Cardiac involvement (50%, HF, pericarditis, arrhythmia) [3]. Eosinophilic myocarditis → myocardial dysfunction. Endomyocardial fibrosis → restrictive physiology |
| Cardiac MRI (CMR) [18] | Gold standard for non-invasive cardiac assessment. Late gadolinium enhancement (LGE) in subendocardial/transmural distribution (reflecting fibrosis/necrosis). Myocardial oedema on T2-weighted images (active inflammation). Pericardial effusion | CMR detects both active myocarditis (oedema) and chronic fibrosis (LGE). Lake Louise criteria are used for MRI diagnosis of myocarditis. Should be performed in ALL EGPA patients given cardiac involvement is the major cause of death |
| CXR / CT for DAH | Bilateral alveolar opacities (may be diffuse) | If haemoptysis present, consider diffuse alveolar haemorrhage — though more common in MPA/GPA |
| Finding | Interpretation |
|---|---|
| Non-specific ST/T changes | Myocarditis — diffuse myocardial inflammation |
| Ventricular arrhythmias | Eosinophilic infiltration disrupts conduction pathways → re-entrant circuits |
| Heart block (any degree) | Conduction tissue infiltration by eosinophils or granulomas |
| ACS-like ST elevation | Coronary vasculitis or direct myocardial injury mimicking acute MI |
| Finding | Interpretation |
|---|---|
| Axonal neuropathy pattern in multiple non-contiguous named nerve distributions | Confirms mononeuritis multiplex — vasculitis of vasa nervorum → nerve infarction → Wallerian degeneration (axonal loss). Sensorimotor involvement. Asymmetric. Common peroneal nerve most frequently affected (foot drop) |
| Reduced SNAP/CMAP amplitudes with relatively preserved conduction velocities | Axonal pattern (vs demyelinating where conduction velocity drops) — consistent with vascular nerve infarction rather than demyelination |
| Test | Findings | Interpretation |
|---|---|---|
| Spirometry | Obstructive pattern (↓ FEV1/FVC ratio) due to asthma. May have mixed obstructive-restrictive if parenchymal disease | Confirms airway obstruction from bronchospasm. Restrictive component suggests eosinophilic lung parenchymal disease |
| DLCO | ↑ DLCO if diffuse alveolar haemorrhage (carbon monoxide binds to intra-alveolar haemoglobin). ↓ DLCO if interstitial lung disease | Elevated DLCO paradoxically indicates DAH — a useful test when haemoptysis is present |
| Bronchoalveolar lavage (BAL) [17] | Bronchoalveolar lavage shows pulmonary eosinophilia [17]. BAL eosinophils > 25% is consistent with eosinophilic lung disease. In active EGPA, BAL may show 30–80% eosinophils | BAL is useful but not diagnostic in isolation. It confirms eosinophilic alveolitis and helps exclude infection. Important to send for microbiology (AFB, fungal culture) to exclude TB and ABPA |
Biopsy of involved organs: confirm presence of eosinophilic inflammatory process or vasculitis [17]. Sample collections include skin biopsy, sural nerve biopsy, bronchoalveolar lavage [17]
| Biopsy Site | When to Choose | Histological Findings | Interpretation |
|---|---|---|---|
| Skin biopsy (most accessible) | Palpable purpura, subcutaneous nodules | Haemorrhagic lesions show leukocytoclastic vasculitis [17]. Vascular lesions show granuloma, eosinophilic infiltrates and fibrinoid necrosis [17] | Leukocytoclastic vasculitis = neutrophilic debris ("nuclear dust") around small dermal vessels with fibrinoid necrosis. In EGPA, the eosinophilic component is prominent |
| Sural nerve biopsy | Mononeuritis multiplex with lower limb involvement | Sural nerve biopsy shows necrotizing granuloma which is mainly histiocytes and multinucleated giant cells surrounding degenerated collagen fibres [17]. Vasculitis of epineurial/perineurial vessels with eosinophilic infiltration | Demonstrates vasculitis of vasa nervorum directly. The sural nerve is chosen because it is a pure sensory nerve in the calf — biopsy causes only a small sensory deficit on the lateral foot |
| Renal biopsy | Haematuria, proteinuria, rising creatinine (suspected GN) | Pauci-immune focal segmental necrotizing and crescentic GN (Type III RPGN). Minimal staining on immunofluorescence (pauci-immune) [13]. Eosinophilic interstitial infiltration may be present | Immunofluorescence pattern most helpful for diagnosis [12]. Pauci-immune IF (negative/minimal staining) distinguishes ANCA-associated GN from immune complex GN (granular) and anti-GBM disease (linear) |
| Lung biopsy (transbronchial or surgical) | Pulmonary infiltrates, diagnostic uncertainty | Eosinophilic necrotizing granulomas, small vessel vasculitis, eosinophilic pneumonia | Most specific but most invasive. Transbronchial biopsy may have limited yield due to small sample size |
| Endomyocardial biopsy | Suspected eosinophilic myocarditis not responding to treatment | Eosinophilic myocardial infiltration, myocyte necrosis, fibrosis | Gold standard for myocarditis diagnosis but associated with risk of cardiac perforation (1%) [18]. Usually reserved for cases where diagnosis will change management |
Histology — The Three Classic Findings
Eosinophilic necrotising granuloma [3][6]: the pathological hallmark of EGPA. The three key histological features are:
- Eosinophilic infiltrates — eosinophils surrounding and infiltrating vessel walls and tissues
- Necrotizing vasculitis — fibrinoid necrosis of small vessel walls
- Granulomatous inflammation — epithelioid histiocytes and multinucleated giant cells forming granulomas, often with central eosinophilic necrosis ("allergic granuloma")
IF pattern: Pauci-immune (minimal/absent Ig and complement deposition) — this is the hallmark of ANCA-associated vasculitis and distinguishes it from immune-complex vasculitides.
3I. Disease Activity and Severity Assessment
- A validated scoring system used for all AAVs to quantify disease activity
- Scores organ involvement across 9 domains (general, cutaneous, mucous membranes/eyes, ENT, chest, cardiovascular, abdominal, renal, nervous system)
- Higher score = more active disease
- Used to guide treatment intensity and monitor response
The Five-Factor Score (FFS) is used to assess prognosis and guide treatment intensity in EGPA. Developed by the French Vasculitis Study Group (FVSG):
| Factor | Points | Why It's a Bad Prognostic Factor |
|---|---|---|
| Cardiac involvement | +1 | Major cause of death — eosinophilic myocarditis |
| GI involvement | +1 | Mesenteric vasculitis → bowel ischaemia, perforation |
| Renal insufficiency (Cr > 150 µmol/L) | +1 | Indicates significant renal vasculitis / RPGN |
| Proteinuria > 1 g/day | +1 | Indicates significant glomerular damage |
| CNS involvement | +1 | Cerebral vasculitis (rare but severe) |
- FFS = 0: Good prognosis; may be treated with steroids alone
- FFS ≥ 1: Poor prognosis; requires steroids + cyclophosphamide or rituximab
| Investigation | Purpose | Expected Result in EGPA |
|---|---|---|
| Stool O&P, Strongyloides serology | Exclude parasitic eosinophilia | Negative |
| Aspergillus skin test, specific IgE to Aspergillus | Exclude ABPA | Negative (or if weakly positive, total IgE not as high as ABPA) |
| FIP1L1-PDGFRA fusion (FISH/PCR) | Exclude clonal hypereosinophilic syndrome | Negative |
| Peripheral blood smear, bone marrow biopsy | Exclude eosinophilic leukaemia / myeloproliferative neoplasm | No blast cells, no clonal eosinophilic proliferation |
| Drug history review | Exclude drug-induced eosinophilia | Temporal relationship absent |
| Tryptase level | Exclude mastocytosis (which can co-exist with eosinophilia) | Normal |
| Phase | Investigations | Purpose |
|---|---|---|
| First line | CBC D/C, ↑↑ ESR/CRP, ANCA, RFT & urinalysis [1] | Confirm eosinophilia, inflammation, screen for renal involvement, ANCA status |
| Serological panel | p-ANCA/anti-MPO, c-ANCA/anti-PR3, ANA, anti-dsDNA, anti-GBM, complement C3/C4, total IgE | Narrow DDx: AAV vs immune complex vs anti-GBM vs ABPA |
| Organ assessment | CXR/HRCT, Echo/CMR, ECG, NCS/EMG, CT sinuses, urinalysis | Map organ involvement (lungs, heart, nerves, sinuses, kidneys) |
| Tissue biopsy | Skin biopsy: histology & direct IF; biopsy of involved organs: kidney, vessels [1] | Confirm eosinophilic necrotizing granuloma and pauci-immune pattern |
| Exclude mimics | Stool O&P, Strongyloides/Aspergillus serology, FIP1L1-PDGFRA, drug review | Rule out parasites, ABPA, HES, drugs |
| Severity | FFS score, BVAS, cardiac biomarkers, CMR | Guide treatment intensity |
High Yield Summary — Diagnosis of EGPA
ACR 1990 Criteria: ≥ 4/6 of: asthma, eosinophilia > 10%, mononeuropathy/polyneuropathy, migratory pulmonary infiltrates, paranasal sinus abnormality, extravascular eosinophils on biopsy.
2022 ACR/EULAR Criteria: Weighted scoring; eosinophil count ≥ 1 × 10⁹/L carries +5 points; positive c-ANCA/anti-PR3 is a NEGATIVE criterion (−3) because it suggests GPA.
Key investigations: CBC D/C (eosinophilia > 10%), ESR/CRP (↑↑), ANCA (p-ANCA/anti-MPO in ~40–50%), complement (normal — rules out IC disease), urinalysis, CXR/HRCT, Echo/CMR, NCS/EMG, tissue biopsy.
Biopsy: Eosinophilic necrotising granuloma + necrotising vasculitis + pauci-immune IF. Skin biopsy is most accessible; sural nerve biopsy in mononeuritis multiplex; renal biopsy if GN suspected.
FFS: Cardiac, GI, renal insufficiency (Cr > 150), proteinuria > 1g/d, CNS. FFS ≥ 1 → add cyclophosphamide.
ANCA negative does NOT exclude EGPA — 50–60% are ANCA-negative.
Active Recall - Diagnosis and Investigations of EGPA
References
[1] Senior notes: Maksim Medicine Notes.pdf (Rheumatology — ANCA-associated vasculitis, Investigations, p.331) [2] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (Rheumatological Diseases — EGPA overview, CHCC definition, pp.1764–1769) [3] Senior notes: Ryan Ho Rheumatology.pdf (Vasculitis — AAV comparison table, pp.93–97) [6] Lecture slides: GC 053. Fingers turn white and blue.pdf (p.94 — EGPA vs GPA comparison table) [9] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (Asthma-associated syndromes — ABPA criteria, p.188) [12] Senior notes: Ryan Ho Fundamentals.pdf (Nephritic syndrome evaluation — complement levels, serology, pp.359–361) [13] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (RPGN classification by IF pattern, pp.413–421) [15] Senior notes: Ryan Ho Urogenital.pdf (ANCA-associated GN — diagnosis: ANCA suggestive but not diagnostic, p.69) [17] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (EGPA — Diagnosis: biochemical tests, biopsy, pp.1769–1771) [18] Senior notes: Ryan Ho Cardiology.pdf (Myocarditis — evaluation and management, p.165)
Management of EGPA (Churg-Strauss Syndrome)
Management of EGPA follows the same overarching framework used for all ANCA-associated vasculitides (AAV): a two-phase approach of (1) induction of remission to rapidly control active vasculitis and organ damage, followed by (2) maintenance of remission with less toxic agents to prevent relapse. What makes EGPA management unique compared to GPA/MPA is:
- The Five-Factor Score (FFS) drives treatment intensity (rather than the ANCA titre or organ-specific criteria used in GPA/MPA)
- Corticosteroids alone may suffice for mild/non-organ-threatening disease (FFS = 0) — a key difference from GPA/MPA where cyclophosphamide or rituximab is almost always needed
- Concurrent asthma management must continue throughout — the patient never stops being asthmatic
- Cardiac involvement requires aggressive treatment because it is the leading cause of death
- Mepolizumab (anti-IL-5) has become a game-changer for relapsing/refractory EGPA — the first biologic specifically licensed for EGPA (FDA-approved 2017)
3. Treatment Modalities — Detailed Breakdown
3A. Induction of Remission
Induction of remission: High-dose corticosteroids + Cyclophosphamide [17]
Why corticosteroids work in EGPA: Corticosteroids ("gluco-cortico-steroids" — "gluco" = glucose metabolism, "cortico" = adrenal cortex, "steroid" = cholesterol-derived structure) are the most effective agents because they:
- Suppress Th2 cytokine production (IL-4, IL-5, IL-13) → reduces eosinophil differentiation and survival
- Induce eosinophil apoptosis directly — cortisol activates pro-apoptotic pathways in eosinophils (unlike neutrophils, where steroids prolong survival)
- Suppress NF-κB → reduces transcription of inflammatory mediators (TNF-α, IL-1, adhesion molecules)
- Reduce ANCA production indirectly by suppressing B-cell activation
| Regimen | Indication | Details |
|---|---|---|
| Oral prednisolone 0.5–1 mg/kg/day | FFS = 0 (non-severe EGPA) or after IV pulse | Start at 1 mg/kg/day (max 60–80 mg/day). Maintain for 3–4 weeks until clinical response, then taper gradually over 9–12 months |
| IV pulse methylprednisolone 1 g/day × 3 days [19] | FFS ≥ 1 (severe EGPA) with organ-threatening disease (cardiac, renal, neurological) | Requires pulse steroids (IV methylprednisolone 1 g/day for 3 days) [19]. The rationale for IV pulse is to achieve rapid, supraphysiological immunosuppression before oral therapy takes effect. Follow with oral prednisolone 1 mg/kg/day |
Steroid taper: This is one of the most challenging aspects of EGPA management. Unlike GPA/MPA, EGPA patients often cannot be fully weaned off steroids because:
- The underlying asthma requires ongoing corticosteroid therapy
- EGPA has a high relapse rate when steroids are withdrawn
- Typical target: taper to prednisolone 5–7.5 mg/day as maintenance (some patients require lifelong low-dose steroids)
Why Can't We Just Stop Steroids?
EGPA patients are uniquely steroid-dependent compared to GPA/MPA patients. Their asthma worsens when steroids are tapered, and the vasculitis may relapse. This is why steroid-sparing agents are essential — to allow steroid dose reduction while maintaining remission. It is also why LTRA can unmask EGPA [5] — the LTRA provides asthma control, encouraging clinicians to taper steroids, which then unmasks the vasculitic disease.
Contraindications / Cautions for corticosteroids:
- Uncontrolled diabetes mellitus (steroids cause hyperglycaemia via gluconeogenesis stimulation and peripheral insulin resistance)
- Active infection (steroids suppress immune surveillance — must exclude TB, Strongyloides before starting)
- Osteoporosis (long-term use → osteoblast suppression, osteoclast activation)
- Peptic ulcer disease (steroids reduce prostaglandin-mediated gastroprotection)
- Psychosis (rare but dose-dependent)
Induction: high-dose steroid ± cyclophosphamide [3]
Name breakdown: "Cyclo" = ring structure, "phosph" = phosphorus-containing, "amide" = nitrogen-linked. It's a nitrogen mustard alkylating agent — a chemotherapy drug repurposed as an immunosuppressant.
Mechanism: CYC is a prodrug metabolised in the liver to its active metabolites (phosphoramide mustard + acrolein). Phosphoramide mustard:
- Cross-links DNA strands → prevents DNA replication → kills rapidly dividing cells (lymphocytes, especially B cells)
- Depletes B cells → reduces ANCA production and immune complex formation
- Depletes T cells → reduces Th2 cytokine drive
Indication in EGPA: FFS ≥ 1 (cardiac, GI, renal, CNS involvement, or proteinuria > 1 g/day)
| Route | Regimen | Advantages | Disadvantages |
|---|---|---|---|
| IV pulse (preferred) | 15 mg/kg (max 1.2 g) every 2–3 weeks × 6 pulses | Lower cumulative dose → fewer side effects (especially bladder toxicity and infertility) | Requires hospital attendance |
| Oral daily | 2 mg/kg/day for 3–6 months | Continuous exposure → may be more effective in very severe disease | Higher cumulative dose → more side effects |
Important side effects of cyclophosphamide:
| Side Effect | Mechanism | Prevention |
|---|---|---|
| Haemorrhagic cystitis | Acrolein (toxic metabolite) is excreted in urine → damages bladder urothelium | Mesna (2-mercaptoethane sodium sulfonate) — binds acrolein in urine, neutralising it. Also ensure good hydration (dilutes acrolein concentration) |
| Bone marrow suppression | Kills rapidly dividing haematopoietic progenitors → neutropenia, thrombocytopenia | Monitor FBC regularly (every 2 weeks during induction). Adjust dose to nadir neutrophil count > 1.5 × 10⁹/L |
| Infertility | Gonadal toxicity — alkylates oocytes and spermatogonia | Discuss sperm/oocyte cryopreservation before starting. Consider GnRH agonist to suppress ovarian function during treatment |
| Infections (major concern) | Profound immunosuppression → opportunistic infections (PJP, herpes zoster, CMV) | Co-trimoxazole prophylaxis against PJP (trimethoprim-sulfamethoxazole 480 mg daily or 960 mg thrice weekly) |
| Malignancy (long-term) | Alkylation → DNA damage → ↑ risk of bladder carcinoma, lymphoma, MDS/AML | Minimise cumulative dose; switch to maintenance agent as soon as remission achieved |
| Nausea/vomiting | Direct GI mucosal irritation + CTZ stimulation | Anti-emetics (ondansetron) before IV pulses |
Contraindications: Active severe infection, pregnancy (teratogenic — category D), severe pre-existing cytopenias, bladder pathology (prior haemorrhagic cystitis)
Name breakdown: Rituximab = "ri-tuxi-mab" — "-mab" = monoclonal antibody; originally developed as a chimeric anti-CD20 antibody.
Mechanism: Rituximab targets CD20 on the surface of pre-B cells and mature B cells → complement-dependent cytotoxicity (CDC) + antibody-dependent cellular cytotoxicity (ADCC) + direct apoptosis → B cell depletion → reduced ANCA production, reduced immune-complex formation.
Why use it in EGPA?
- Effective alternative to CYC, particularly in patients who cannot tolerate CYC (e.g., young women concerned about fertility)
- RAVE and RITUXVAS trials demonstrated non-inferiority of rituximab to CYC for AAV induction (these trials primarily enrolled GPA/MPA patients, but data is extrapolated to EGPA)
- Particularly useful in ANCA-positive EGPA where the vasculitic phenotype is driven by B cell–mediated ANCA production
- Less robust data in ANCA-negative EGPA where the eosinophilic/tissue-infiltrative pathway dominates
| Regimen | Details |
|---|---|
| Standard induction | 375 mg/m² IV weekly × 4 weeks (lymphoma protocol) OR 1 g IV × 2 doses 2 weeks apart (RA protocol) |
| Pre-medication | IV methylprednisolone, paracetamol, antihistamine (to reduce infusion reactions) |
Side effects: Infusion reactions (fever, rigors, hypotension), severe infections (hypogammaglobulinaemia from sustained B cell depletion), progressive multifocal leukoencephalopathy (PML — very rare; caused by JC virus reactivation), hepatitis B reactivation (must screen HBsAg/anti-HBc before starting)
Contraindications: Active severe infection, HBV reactivation risk without prophylaxis, pregnancy/breastfeeding
Once remission is achieved (typically after 3–6 months of induction), the goal is to switch from CYC to a less toxic steroid-sparing agent while continuing low-dose prednisolone.
Maintenance of remission: Low-dose corticosteroids + Azathioprine / Leflunomide / Methotrexate [17]
Maintenance: azathioprine / methotrexate / leflunomide [3]
| Agent | Mechanism | Dose | Key Considerations |
|---|---|---|---|
| Azathioprine (AZA) | Purine analogue → inhibits DNA synthesis in lymphocytes. Metabolised to 6-mercaptopurine (6-MP) by hypoxanthine-guanine phosphoribosyltransferase (HGPRT) → incorporated into DNA → chain termination | 2 mg/kg/day orally | Check TPMT genotype before starting — thiopurine methyltransferase (TPMT) metabolises AZA; homozygous TPMT deficiency → severe myelosuppression. S/E: bone marrow suppression, hepatotoxicity, pancreatitis, GI upset, skin cancer risk. Avoid allopurinol (inhibits xanthine oxidase, an alternative pathway for AZA metabolism → severe toxicity) |
| Methotrexate (MTX) | Dihydrofolate reductase (DHFR) inhibitor → blocks purine and thymidylate synthesis → suppresses lymphocyte proliferation. Also has anti-inflammatory effects via adenosine accumulation | 15–25 mg weekly (oral or SC) | Requires folic acid supplementation (5 mg next day) to reduce mucosal and marrow toxicity. S/E: hepatotoxicity (monitor LFTs), myelosuppression, pneumonitis (a rare but dangerous complication — must differentiate from EGPA lung disease!), teratogenic (absolute contraindication in pregnancy). Avoid in significant renal impairment (renally cleared → accumulation) |
| Leflunomide | Inhibits dihydroorotate dehydrogenase (DHODH) → blocks de novo pyrimidine synthesis → suppresses T and B lymphocyte proliferation | 20 mg daily | S/E: hepatotoxicity (monitor LFTs), diarrhoea, alopecia, teratogenic (very long half-life — if pregnancy desired, requires washout with cholestyramine). Less commonly used than AZA or MTX in Hong Kong practice |
| Mycophenolate mofetil (MMF) | Inhibits inosine monophosphate dehydrogenase (IMPDH) → blocks de novo purine synthesis → selectively suppresses lymphocytes (which depend on de novo pathway, unlike other cells that can use salvage pathway) | 1–1.5 g BD | Alternative option. S/E: GI upset (diarrhoea, nausea), myelosuppression, teratogenic. Less evidence base in EGPA than AZA/MTX but used in refractory cases |
Duration of maintenance: Continue for 18–24 months minimum [1], often longer in EGPA due to high relapse rates. Some patients require lifelong maintenance.
Low-dose prednisolone: Continue at 5–7.5 mg/day during maintenance. Attempt further tapering every 3–6 months guided by clinical status, eosinophil count, and inflammatory markers.
Why Switch from CYC to Maintenance Agents?
Cyclophosphamide is too toxic for long-term use (cumulative dose-dependent bladder cancer risk, infertility, myelosuppression). The principle is: use CYC as a "big gun" to put out the fire (induction), then switch to a "lower-calibre" drug to keep the fire out (maintenance). This is analogous to the concept of loading dose → maintenance dose in pharmacology.
3C. Biologic Therapies — The Modern Era
Name breakdown: Mepolizumab — "-mab" = monoclonal antibody; "mepo" is a proprietary prefix; targets IL-5.
Mechanism: Mepolizumab is a humanised monoclonal anti-IL-5 antibody. IL-5 is the key cytokine driving:
- Eosinophil differentiation from bone marrow precursors
- Eosinophil maturation, activation, and survival
- Eosinophil tissue migration
By blocking IL-5, mepolizumab:
- Reduces peripheral blood eosinophil count
- Reduces eosinophilic tissue infiltration (heart, lungs, GI)
- Reduces EGPA relapse rates
- Allows steroid dose reduction (steroid-sparing)
Evidence: The MIRRA trial (2017) demonstrated that mepolizumab 300 mg SC every 4 weeks significantly:
- Increased proportion of patients in remission
- Reduced relapse rates
- Allowed glucocorticoid dose reduction
compared to placebo in relapsing/refractory EGPA.
Indication:
- Relapsing EGPA — patients who relapse when steroids are tapered
- Refractory EGPA — patients not responding adequately to steroids ± conventional immunosuppressants
- Steroid-sparing — to facilitate steroid tapering in steroid-dependent patients
- FDA-approved and EMA-approved for EGPA at 300 mg SC every 4 weeks (note: the dose for severe eosinophilic asthma is only 100 mg; EGPA requires the higher 300 mg dose)
Side effects: Injection site reactions, headache, back pain, fatigue. Generally well tolerated. Rare: herpes zoster reactivation (eosinophils play a minor role in antiviral defence), hypersensitivity reactions.
Contraindications: Known hypersensitivity to mepolizumab. Pre-existing helminthic infection should be treated before starting (eosinophils are crucial for anti-helminth defence — depleting them may allow parasite dissemination, particularly Strongyloides hyperinfection).
Mepolizumab for EGPA vs Asthma — Dose Matters
The dose of mepolizumab for EGPA (300 mg SC Q4W) is three times the dose used for severe eosinophilic asthma (100 mg SC Q4W). This is because EGPA involves both tissue eosinophilia and systemic vasculitis, requiring greater IL-5 suppression than airway eosinophilia alone.
Mechanism: Benralizumab targets the IL-5 receptor α-chain (IL-5Rα) directly on eosinophils → not only blocks IL-5 signalling but also induces eosinophil apoptosis via enhanced ADCC (afucosylated Fc region enhances NK cell binding). This results in near-complete eosinophil depletion.
Status in EGPA: The MANDARA trial (2023–2024) showed non-inferiority of benralizumab to mepolizumab in EGPA. Benralizumab received FDA approval for EGPA in 2024. It offers the advantage of deeper eosinophil depletion and less frequent dosing (30 mg SC Q8W after initial loading).
Rituximab can also be used as a maintenance agent (500 mg IV every 6 months), particularly in ANCA-positive EGPA with vasculitic phenotype. Data is more limited in EGPA compared to GPA/MPA but growing.
This comparison from the senior notes is extremely high yield [3]:
| MPA | EGPA | GPA | |
|---|---|---|---|
| Induction | High-dose steroid + cyclophosphamide / rituximab | High-dose steroid ± cyclophosphamide | High-dose steroid + cyclophosphamide / rituximab (± plasma exchange if severe) |
| Maintenance | AZA / RIT / MTX | Azathioprine / methotrexate / leflunomide | Azathioprine / rituximab / methotrexate |
| Prognosis | 4–10y survival 56–95%. Lung/renal and infection as major cause of death | 5y survival 70–90%. Cardiac as major cause of death | 4–10y survival 56–95%. ↑ relapse risk cf GPA. Lung/renal and infection as major cause of death |
The key difference: EGPA is the only AAV where steroids alone may suffice for mild disease (FFS = 0), and CYC is added only if needed ("±" rather than "+"). In GPA and MPA, CYC or rituximab is virtually always part of induction.
4. Concurrent and Supportive Management
EGPA patients remain asthmatic for life. Asthma management runs in parallel with vasculitis management:
- Inhaled corticosteroids (ICS) ± long-acting β₂-agonists (LABA): Mainstay of asthma control. Continue as per standard GINA stepwise approach
- Avoid over-reliance on LTRA: Leukotriene receptor antagonists can unmask previously undiagnosed Churg-Strauss Syndrome [5][20]. In known EGPA, LTRAs are not contraindicated per se, but the clinician must be aware that steroid tapering after adding LTRA may unmask vasculitic relapse
- Anti-IL-5 therapy (mepolizumab/benralizumab): Serves dual purpose — controls both eosinophilic asthma AND EGPA vasculitis
Churg-Strauss syndrome is an ANCA-related vasculitis. It may present as asthma in its early stages but may progress to widespread eosinophilic vasculitic damage to various organs. It is commonly mis-diagnosed as asthma and coincidentally can be treated by ICS. When such patient switches from steroids to LTRA, systemic manifestations will resurface. [5]
Cardiac involvement is a common cause of morbidity and mortality [2]:
| Complication | Management |
|---|---|
| Eosinophilic myocarditis | Immunosuppression in known autoimmune disease, giant cell or non-infectious eosinophilic myocarditis [18]. Aggressive induction with IV pulse methylprednisolone + CYC. DON'T give NSAIDs (↓ PG production → may worsen myocardial function + ↑ myocardial necrosis) [18] |
| Heart failure | Standard HF management: ACE inhibitors/ARBs, beta-blockers, diuretics, MRA. Treat heart failure and arrhythmia ± organ support [18] |
| Arrhythmias | Antiarrhythmic drugs as per type. Consider ICD if high risk of sudden cardiac death |
| Intracardiac thrombus | Anticoagulation (eosinophilic endocardial damage creates a prothrombotic surface) |
| Pericardial effusion | Usually responds to systemic steroids. Pericardiocentesis if haemodynamic compromise (tamponade) |
- Monitor RFT and urinalysis regularly
- If RPGN develops: treat as per AAV-GN — pulse steroids + cyclophosphamide (or rituximab) ± plasma exchange [19]
- Plasma exchange: Consider if severe renal failure (Cr > 500 µmol/L) or diffuse alveolar haemorrhage (removes circulating ANCA and inflammatory mediators). The PEXIVAS trial showed limited benefit of plasma exchange for renal outcomes in AAV, but it may still be considered in life-threatening situations
- Immunosuppressive treatment of the vasculitis itself is the primary treatment (nerve recovery follows vasculitis control)
- Pain management: Neuropathic pain agents — pregabalin, gabapentin, amitriptyline, duloxetine
- Physiotherapy / Occupational therapy: For foot drop (ankle-foot orthosis), wrist drop (wrist splint), rehabilitation
- Recovery is slow (months to years) and may be incomplete if axonal loss is severe
All patients on immunosuppressive therapy (especially high-dose steroids + CYC/RTX) need:
| Prophylaxis | Agent | Rationale |
|---|---|---|
| PJP prophylaxis | Co-trimoxazole (trimethoprim-sulfamethoxazole) 480 mg daily or 960 mg three times weekly | Pneumocystis jirovecii pneumonia is a devastating opportunistic infection in immunosuppressed patients. Standard prophylaxis during induction and maintained while on significant immunosuppression |
| Strongyloides screening | Serology before starting immunosuppression | Strongyloides hyperinfection syndrome can occur when immunosuppression (especially steroids) leads to unopposed larval proliferation. Particularly relevant in Hong Kong (SE Asian migration) |
| HBV screening | HBsAg, anti-HBc before rituximab | HBV reactivation can be fatal. If positive, antiviral prophylaxis (entecavir/tenofovir) required |
| Vaccination | Influenza (annual), pneumococcal, COVID-19. Avoid live vaccines | Immunosuppressed patients are at ↑ risk of infections. Live vaccines (MMR, varicella, BCG, oral typhoid) are contraindicated as they could cause active infection |
| Herpes zoster prophylaxis | Consider aciclovir/valaciclovir if high risk | Particularly with rituximab or CYC |
All patients on prednisolone ≥ 7.5 mg/day for ≥ 3 months need:
- Calcium supplementation (1000–1200 mg/day) + Vitamin D (800–1000 IU/day)
- Bisphosphonate (alendronate 70 mg weekly or risedronate 35 mg weekly) — particularly in postmenopausal women and men > 50 years
- DEXA scan at baseline and periodically
- Proton pump inhibitor (omeprazole, pantoprazole) if on high-dose steroids ± concurrent NSAID or anticoagulant use
- Rationale: Steroids reduce gastric mucosal prostaglandin synthesis → reduced gastroprotection → increased ulcer risk
- Steroids cause metabolic syndrome (hyperglycaemia, hypertension, dyslipidaemia)
- Regular monitoring of blood glucose, blood pressure, lipid profile
- Lifestyle advice: Diet, exercise (as tolerated), smoking cessation
EGPA has a high relapse rate — approximately 25–50% of patients relapse within 5 years. Relapse can be:
- Minor relapse: Worsening asthma, rising eosinophils, mild constitutional symptoms → increase prednisolone dose, consider adding/changing maintenance agent
- Major relapse: New organ-threatening disease (cardiac, renal, neurological) → re-induce with pulse steroids + CYC or rituximab
Mepolizumab is the preferred option for relapsing or steroid-dependent EGPA, as it directly targets the IL-5/eosinophil axis and has the strongest evidence base in this setting.
| Parameter | Frequency | Purpose |
|---|---|---|
| CBC with eosinophil count | Every 2–4 weeks during induction; every 1–3 months during maintenance | Monitor eosinophil response (should fall with treatment), detect myelosuppression from CYC/AZA/MTX |
| ESR, CRP | Every visit | Track inflammatory activity |
| RFT, LFT | Every visit | Monitor renal function and hepatotoxicity from MTX/AZA |
| Urinalysis | Every visit | Detect new/worsening GN (haematuria, proteinuria) |
| ANCA titre | Every 3–6 months (in ANCA-positive patients) | Rising ANCA may predict relapse (though not used in isolation to guide treatment changes) |
| Cardiac imaging | Echo at baseline and as indicated; CMR if cardiac involvement | Monitor cardiac function; cardiac involvement is the major cause of death |
| Spirometry | Every 6–12 months | Monitor asthma control |
| NCS/EMG | As clinically indicated | Monitor neuropathy recovery |
5-year survival 70–90%. Cardiac as major cause of death [3]
- Before corticosteroid era: 5-year survival was approximately 25%
- With modern treatment: 5-year survival improved to 70–90%
- FFS = 0: 5-year mortality < 10%
- FFS ≥ 2: 5-year mortality up to 40%
- Main causes of death: Eosinophilic myocarditis / heart failure (most common), infection (from immunosuppression), GI perforation from mesenteric vasculitis
- Relapse: 25–50% relapse within 5 years; asthma never fully remits
High Yield Summary — Management of EGPA
Treatment framework: Induction → Maintenance, guided by Five-Factor Score (FFS).
FFS = 0 (non-severe): High-dose prednisolone alone (1 mg/kg/day, taper over 9–12 months).
FFS ≥ 1 (severe): IV pulse methylprednisolone (1 g/day × 3 days) + cyclophosphamide or rituximab.
Maintenance: Azathioprine / methotrexate / leflunomide + low-dose prednisolone for ≥ 18–24 months.
Biologic: Mepolizumab 300 mg SC Q4W — first-line for relapsing/refractory EGPA (blocks IL-5 → reduces eosinophil count). Benralizumab (anti-IL-5Rα) approved 2024.
EGPA vs GPA/MPA treatment: EGPA is the only AAV where steroids alone may suffice (FFS = 0); CYC is "±" not "+".
Key differences from GPA/MPA: CYC used selectively; cardiac management is critical; mepolizumab has specific role; asthma management is concurrent and lifelong.
Supportive care: PJP prophylaxis (co-trimoxazole), bone protection (calcium + vitamin D + bisphosphonate), HBV screening before rituximab, Strongyloides screening, gastroprotection.
Do NOT give NSAIDs in eosinophilic myocarditis — may worsen myocardial function.
Prognosis: 5-year survival 70–90%; cardiac death is the leading cause.
Active Recall - Management of EGPA
References
[1] Senior notes: Maksim Medicine Notes.pdf (Rheumatology — PAN and ANCA-associated vasculitis management, p.331) [2] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (EGPA overview — cardiac involvement, pp.1767–1769) [3] Senior notes: Ryan Ho Rheumatology.pdf (AAV comparison table — treatment and prognosis, pp.93–97) [5] Senior notes: Ryan Ho Respiratory.pdf (LTRA and Churg-Strauss, footnote 73, p.106); Lecture slides: GC 040. Cough and wheezing_asthma and allergic lung diseases.pdf (p.40) [17] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (EGPA — Treatment: induction and maintenance, pp.1769–1771) [18] Senior notes: Ryan Ho Cardiology.pdf (Myocarditis — evaluation and management, p.165) [19] Senior notes: MBBS Final MB (Pediatrics) (Felix PY Lai).pdf (ANCA+ve vasculitis — pulse steroids + CYC/rituximab, p.413) [20] Senior notes: Adrian Lui Pediatrics Notes.pdf (LTRA — can unmask Churg-Strauss, footnote 111, p.175)
Complications of EGPA (Churg-Strauss Syndrome)
Complications of EGPA can be divided into two broad categories: (A) Disease-related complications — the direct consequences of eosinophilic infiltration and vasculitis on target organs; and (B) Treatment-related complications — the iatrogenic harm from prolonged immunosuppression, corticosteroids, and cytotoxic agents. Understanding both is essential because in EGPA, as in all AAVs, the treatment itself can be as dangerous as the disease.
1. Disease-Related Complications — Organised by Organ System
Major cause of death in EGPA = Cardiac [6] Cardiac involvement is a common cause of morbidity and mortality [2] Cardiac (50%, HF, pericarditis, arrhythmia) [3]
Cardiac complications account for roughly 50% of EGPA-related deaths and are the single most important differentiator from GPA and MPA (where pulmonary/renal disease and infection dominate mortality). Cardiac disease is more common in the ANCA-negative phenotype, where eosinophilic tissue infiltration predominates.
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Eosinophilic myocarditis | Eosinophils infiltrate the myocardium and degranulate → release of major basic protein (MBP), eosinophil cationic protein (ECP), and eosinophil peroxidase (EPO) → direct cytotoxicity to cardiomyocytes → myocardial necrosis. This is followed by fibroblast activation → endomyocardial fibrosis | Acute phase: ↑ troponin, chest pain, dyspnoea, arrhythmias, acute heart failure, cardiogenic shock. Chronic phase: restrictive cardiomyopathy (rigid, fibrosed ventricles that cannot fill properly during diastole → elevated filling pressures → congestion) |
| Heart failure (HF) | (1) Acute myocarditis → systolic dysfunction (dilated cardiomyopathy). (2) Chronic endomyocardial fibrosis → diastolic dysfunction (restrictive cardiomyopathy). Both lead to reduced cardiac output and backward failure | Dyspnoea, orthopnoea, PND, peripheral oedema, elevated JVP, hepatomegaly, ascites. Treat heart failure and arrhythmia ± organ support [18] |
| Arrhythmias | Eosinophilic infiltration of the conduction system (SA node, AV node, His-Purkinje) → disruption of normal electrical pathways. Also, myocardial scarring creates re-entrant circuits | Ventricular tachycardia (VT), ventricular fibrillation (VF), complete heart block, atrial fibrillation. May cause sudden cardiac death — the most feared complication |
| Pericarditis / Pericardial effusion | Eosinophilic infiltration of the pericardium → inflammation and exudate formation | Pleuritic chest pain relieved by sitting forward, pericardial friction rub. Large effusion → cardiac tamponade (Beck's triad: hypotension, distended neck veins, muffled heart sounds) |
| Intracardiac thrombus | Damaged endocardium (from eosinophilic endocarditis — Löffler endocarditis) becomes a prothrombotic surface. Eosinophil granule proteins are also procoagulant (MBP activates factor XII and inhibits thrombomodulin) | Mural thrombus, typically in the ventricular apex → risk of systemic thromboembolism (stroke, peripheral arterial occlusion, mesenteric ischaemia). Requires anticoagulation |
| Coronary vasculitis | Vasculitis of the coronary arteries → luminal narrowing or thrombosis | Myocardial infarction (MI) pattern — may mimic acute coronary syndrome. Coronary angiography may show focal stenoses or aneurysmal dilatation |
Cardiac Complications — The Eosinophilic Cascade
Think of cardiac damage in EGPA as a three-stage process, analogous to the "Löffler endocarditis" seen in hypereosinophilic syndrome:
Stage 1 — Acute necrotic phase: Eosinophil infiltration → degranulation → cardiomyocyte death → acute myocarditis (↑ troponin, arrhythmias, HF).
Stage 2 — Thrombotic phase: Damaged endocardium becomes thrombogenic → mural thrombus formation → embolic risk.
Stage 3 — Fibrotic phase: Fibroblast proliferation replaces necrotic tissue → endomyocardial fibrosis → restrictive cardiomyopathy → chronic HF.
Early aggressive immunosuppression (pulse methylprednisolone + CYC) aims to halt this cascade at Stage 1 before irreversible fibrosis develops.
Why cardiac complications are especially prominent in ANCA-negative EGPA: In ANCA-negative disease, the dominant pathogenic mechanism is direct eosinophilic tissue infiltration rather than neutrophil-mediated vasculitis. The heart is a prime target for eosinophil homing because damaged endothelium expresses adhesion molecules (VCAM-1, ICAM-1) and eotaxin, which actively recruit eosinophils. Without the "vasculitic" phenotype directing disease to kidneys and nerves, the eosinophilic burden is concentrated in myocardium, lungs, and GI tract.
Mononeuritis multiplex (up to 75%) [3] PNS involvement (70%) esp mononeuritis multiplex [3]
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Mononeuritis multiplex | Vasculitis of the vasa nervorum (the tiny arteries supplying peripheral nerve trunks) → nerve ischaemia → axonal degeneration (Wallerian degeneration). Multiple non-contiguous nerves are affected simultaneously or sequentially because vasculitis strikes randomly across the vascular bed | Foot drop (common peroneal nerve — most frequently affected because its vasa nervorum are vulnerable at the fibular head). Wrist drop (radial nerve). Asymmetric sensory loss, burning neuropathic pain. Recovery is slow (months to years) because axonal regeneration proceeds at only ~1 mm/day, and may be incomplete if axonal loss is severe |
| Distal symmetric polyneuropathy | Widespread small vessel vasculitis affecting multiple vasa nervorum simultaneously → confluent axonal loss mimicking a length-dependent polyneuropathy | Stocking-glove sensory loss, distal weakness. Can evolve from mononeuritis multiplex as individual nerve infarctions coalesce |
| Cranial neuropathy (rare) | Vasculitis of cranial nerve vasa nervorum | Facial nerve palsy (CN VII), hearing loss (CN VIII), diplopia (CN III/IV/VI) |
| CNS vasculitis (rare, < 5%) | Vasculitis of cerebral small vessels → cerebral ischaemia or haemorrhage | Stroke, seizures, encephalopathy. Rare but carries very poor prognosis. One of the five FFS factors |
Renal involvement: 45% [2][6]. ANCA is more frequent when glomerulonephritis is present [2]
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Pauci-immune focal segmental necrotising GN | ANCA (anti-MPO) activates neutrophils → neutrophils adhere to glomerular endothelium → degranulation and ROS release → focal fibrinoid necrosis of glomerular capillary walls → breaks in GBM allow plasma to flood into Bowman's space → macrophage/T-cell influx → crescent formation | Haematuria, proteinuria, rising creatinine. If severe → rapidly progressive GN (RPGN) with crescent formation → can progress to ESRD within days to weeks if untreated |
| Chronic kidney disease (CKD) | After the acute GN episode, even with treatment, some glomeruli undergo irreversible sclerosis → progressive nephron loss → CKD | Gradual decline in eGFR over months to years. May eventually require dialysis |
| Eosinophilic interstitial nephritis | Direct eosinophilic infiltration of the renal interstitium (distinct from glomerular vasculitis) | Can contribute to renal dysfunction independent of GN. May respond to steroids |
Although renal involvement is less common in EGPA (45%) than in GPA (80%) or MPA (90%), when it does occur it can be severe and is associated with the ANCA-positive phenotype.
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Chronic severe asthma | The underlying Th2-driven airway disease never remits even after vasculitis is controlled. Airway remodelling (subepithelial fibrosis, smooth muscle hypertrophy, mucus gland hyperplasia) progresses with each exacerbation | Chronic dyspnoea, frequent exacerbations, steroid dependence. Many patients require lifelong ICS ± LABA ± biologic therapy. This is the complication that most impacts daily quality of life |
| Eosinophilic pneumonia (recurrent) | Eosinophilic infiltration of alveolar parenchyma → alveolar filling → impaired gas exchange. Can recur with disease flares | Cough, fever, dyspnoea, bilateral pulmonary infiltrates on CXR. Usually responds to steroids but may relapse |
| Diffuse alveolar haemorrhage (DAH) (rare in EGPA) | Pulmonary capillaritis → destruction of alveolar capillary walls → blood floods into alveolar spaces | Haemoptysis, hypoxia, bilateral alveolar infiltrates, ↑ DLCO (paradoxical — CO binds to intra-alveolar haemoglobin), anaemia. More common in MPA/GPA but can occur in EGPA |
| Pleural effusion | Eosinophilic pleural inflammation → exudative effusion (often eosinophil-rich) | Dyspnoea, dullness to percussion. Pleural fluid analysis: exudate with high eosinophil percentage |
GI (abd pain) [3]. Eosinophilic infiltrative disease: pneumonia, esophagitis, GE [1]
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Eosinophilic gastroenteritis | Eosinophilic infiltration of the GI mucosa/muscularis/serosa → inflammation, dysmotility, mucosal ulceration | Abdominal pain, nausea, vomiting, diarrhoea, malabsorption, GI bleeding. Can affect any segment from oesophagus to colon |
| Mesenteric vasculitis | Vasculitis of mesenteric arteries → bowel ischaemia | Severe abdominal pain (often post-prandial), bloody diarrhoea. Can progress to bowel infarction and perforation — a surgical emergency with high mortality. This is why GI involvement is one of the 5 FFS factors |
| GI perforation | Full-thickness bowel wall necrosis from ischaemia (mesenteric vasculitis) or transmural eosinophilic infiltration → weakened wall → rupture | Acute peritonitis, septic shock. Requires emergency laparotomy |
| Eosinophilic oesophagitis | Eosinophilic infiltration of oesophageal mucosa → mucosal oedema, stricture formation | Dysphagia, odynophagia, food impaction |
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Digital ischaemia / gangrene | Vasculitis of digital arteries → thrombosis → distal ischaemia | Painful, cyanotic digits → may progress to dry gangrene requiring amputation |
| Skin ulceration | Vasculitis of dermal vessels → skin infarction | Painful ulcers, typically over lower legs; heal slowly; risk of secondary infection |
| Scarring from subcutaneous nodules | Resolution of eosinophilic granulomatous subcutaneous inflammation → fibrosis | Cosmetic concern; tender nodules may persist |
EGPA (and AAV in general) carries an increased risk of venous thromboembolism (VTE):
| Complication | Pathophysiology |
|---|---|
| Deep vein thrombosis (DVT) and pulmonary embolism (PE) | Multifactorial: (1) Endothelial damage from vasculitis → prothrombotic surface. (2) Eosinophil granule proteins (MBP) activate coagulation cascade and inhibit thrombomodulin → hypercoagulability. (3) Systemic inflammation → acute phase reactant elevation (fibrinogen ↑). (4) Immobility from neuropathy-related disability. (5) Nephrotic-range proteinuria → antithrombin III loss |
| Arterial thrombosis | Intracardiac thrombus (as above) → cerebral, mesenteric, or peripheral embolism |
Eosinophils Are Prothrombotic
Eosinophils are often overlooked as contributors to thrombosis. However, eosinophil granule proteins — particularly MBP — directly inhibit thrombomodulin (an endothelial anticoagulant protein), activate factor XII, and stimulate platelet aggregation. This explains the paradoxically high VTE rate in EGPA despite it being primarily an inflammatory condition. Always consider prophylactic or therapeutic anticoagulation in patients with active eosinophilic disease and cardiac involvement.
| Complication | Pathophysiology | Clinical Consequence |
|---|---|---|
| Chronic rhinosinusitis | Persistent eosinophilic mucosal inflammation → mucosal oedema, polyp formation, sinus ostia obstruction | Nasal congestion, anosmia (loss of smell — eosinophilic damage to olfactory epithelium), recurrent sinus infections, facial pain/pressure |
| Recurrent nasal polyposis | Polyps tend to recur after surgical removal because the underlying eosinophilic drive persists | Multiple polypectomies may be required; functional endoscopic sinus surgery (FESS) |
| Serous otitis media / sensorineural hearing loss | ± CSOM, SNHL [3]. Eosinophilic middle ear effusion; cochlear vasculitis → sensorineural hearing loss | Conductive hearing loss from effusion; sensorineural hearing loss from cochlear damage (irreversible) |
2. Treatment-Related Complications
These are the consequences of the medications used to treat EGPA — predominantly corticosteroids and cyclophosphamide.
EGPA patients are often on steroids for years to decades (because the asthma never fully remits and the vasculitis has a high relapse rate), making steroid complications a major source of morbidity.
| Complication | Mechanism | Clinical Consequence |
|---|---|---|
| Steroid-induced osteoporosis | Corticosteroids suppress osteoblast function, promote osteoclast activity, reduce intestinal calcium absorption, and increase renal calcium excretion → negative calcium balance → bone loss | Vertebral compression fractures, hip fractures. Prevention: calcium + vitamin D + bisphosphonate |
| Cushingoid habitus | Cortisol excess → central fat redistribution (visceral adiposity), dorsal fat pad ("buffalo hump"), moon facies, supraclavicular fat pads, striae, skin thinning | Cosmetically distressing; associated with metabolic complications |
| Steroid-induced diabetes mellitus | Corticosteroids stimulate hepatic gluconeogenesis, promote peripheral insulin resistance, and reduce glucose uptake in muscle and fat | Hyperglycaemia requiring monitoring; may need oral hypoglycaemics or insulin |
| Hypertension | Mineralocorticoid effect → sodium and water retention; increased sensitivity to catecholamines | Requires antihypertensive therapy |
| Adrenal suppression | Exogenous corticosteroids suppress HPA axis (negative feedback on ACTH). If steroids are abruptly withdrawn, the adrenals cannot mount an adequate cortisol response → adrenal crisis | Fatigue, hypotension, hypoglycaemia, hyperkalemia. Must taper steroids gradually; stress-dose steroids needed for intercurrent illness/surgery |
| Avascular necrosis (AVN) | Corticosteroids cause fat cell hypertrophy within bone marrow → increased intraosseous pressure → reduced blood flow to the femoral head → bone necrosis | Hip pain, especially with weight-bearing. Bilateral femoral head involvement. May require hip replacement |
| Cataracts (posterior subcapsular) | Corticosteroids alter lens epithelial cell metabolism → posterior subcapsular opacities | Progressive painless visual decline. Requires cataract surgery |
| Glaucoma | Corticosteroids reduce aqueous humour outflow through the trabecular meshwork → ↑ intraocular pressure | Monitor IOP in long-term steroid users |
| Susceptibility to infections | Immunosuppression → impaired cell-mediated immunity and neutrophil function | Increased risk of bacterial, viral (especially herpes zoster), fungal (candidiasis), and parasitic (Strongyloides hyperinfection) infections |
| Myopathy | Corticosteroids cause type II muscle fibre atrophy | Proximal muscle weakness (difficulty rising from chair, climbing stairs). Must differentiate from active EGPA myopathy |
| Psychiatric effects | Corticosteroids alter neurotransmitter levels (serotonin, dopamine) and neuronal excitability | Insomnia, euphoria, depression, psychosis (particularly at high doses) |
| Peptic ulcer disease | Reduced prostaglandin synthesis → reduced gastroprotective mucus and bicarbonate secretion | Epigastric pain, GI bleeding. Prophylaxis with PPI |
| Complication | Mechanism | Clinical Consequence |
|---|---|---|
| Haemorrhagic cystitis | Acrolein (toxic metabolite) excreted in urine → urothelial damage → bleeding | Dysuria, haematuria, bladder pain. Prevention: mesna + hydration |
| Bladder carcinoma (long-term) | Cumulative acrolein-induced urothelial DNA damage → transitional cell carcinoma | Presents with painless haematuria years after CYC exposure. Monitor with urinalysis long-term |
| Gonadal toxicity | Alkylation of oocytes/spermatogonia → premature ovarian failure or azoospermia | Infertility (cumulative dose-dependent). Discuss fertility preservation before treatment |
| Myelosuppression | Alkylation of haematopoietic progenitors → pancytopenia | Neutropenia → infection risk; thrombocytopenia → bleeding risk |
| Secondary malignancy | DNA damage → myelodysplastic syndrome (MDS), acute myeloid leukaemia (AML), non-Hodgkin lymphoma | Years after treatment; risk increases with cumulative dose > 36 g |
Infections are the second leading cause of death in AAV (after the disease itself/cardiac complications in EGPA), largely due to immunosuppressive therapy:
| Infection | Risk Factor | Clinical Significance |
|---|---|---|
| Pneumocystis jirovecii pneumonia (PJP) | High-dose steroids + CYC/RTX | Dry cough, fever, progressive hypoxia, bilateral perihilar GGO on HRCT. Prevented by co-trimoxazole prophylaxis. Must differentiate from EGPA pulmonary infiltrates — PJP typically shows diffuse bilateral GGO rather than the peripheral/migratory pattern of eosinophilic pneumonia |
| Herpes zoster | CYC, rituximab, prolonged steroids | Dermatomal vesicular rash with neuropathic pain. Treat with aciclovir/valaciclovir. Consider prophylaxis |
| Strongyloides hyperinfection | Corticosteroid use in patients with latent Strongyloides infection | Steroids suppress eosinophil-mediated killing of Strongyloides larvae → unopposed autoinfection cycle → disseminated larval migration through lungs, GI tract, CNS → Gram-negative sepsis (larvae carry gut bacteria). Potentially fatal. Screen and treat before immunosuppression |
| HBV reactivation | Rituximab (B cell depletion removes immune surveillance of latent HBV) | Hepatic flare → acute liver failure. Must screen HBsAg and anti-HBc before rituximab; start antiviral prophylaxis if positive |
| Bacterial infections | Neutropenia from CYC; impaired phagocyte function from steroids | Pneumonia, urinary tract infections, skin/soft tissue infections, sepsis |
| Complication | Mechanism |
|---|---|
| Hypogammaglobulinaemia | Sustained B cell depletion → reduced immunoglobulin production → recurrent sinopulmonary infections. Monitor IgG levels; consider IVIG replacement if IgG < 4 g/L with recurrent infections |
| Progressive multifocal leukoencephalopathy (PML) | JC virus reactivation due to severe immunosuppression. Extremely rare but devastating — progressive neurological decline, no effective treatment. MRI shows multifocal white matter lesions without mass effect |
| Late-onset neutropenia | Occurs weeks to months after rituximab; mechanism incompletely understood (possibly related to B cell recovery dynamics affecting granulocyte homeostasis) |
EGPA has one of the highest relapse rates among the AAVs — approximately 25–50% relapse within 5 years even with maintenance therapy.
| Aspect | Details |
|---|---|
| Timing | Most relapses occur during steroid tapering or after discontinuation of maintenance immunosuppression |
| Triggers | Steroid tapering, LTRA substitution for steroids (when such patient switches from steroids to LTRA, systemic manifestations will resurface [5]), non-compliance, infection |
| Patterns | Minor relapse: worsening asthma + rising eosinophils + constitutional symptoms. Major relapse: new organ-threatening disease (cardiac, renal, neurological) |
| Consequences | Each relapse causes cumulative organ damage (more myocardial fibrosis, more nephron loss, more axonal loss). Recurrent relapses → progressive disability and end-organ failure |
| Management | Increase steroid dose ± re-induction with CYC/RTX for major relapse. Consider mepolizumab for relapsing/refractory disease |
| Category | Key Complications | Impact on Mortality | Impact on Morbidity |
|---|---|---|---|
| Cardiac (disease) | Myocarditis, HF, arrhythmias, thromboembolism | #1 cause of death [6] | Chronic HF, steroid-dependent cardiomyopathy |
| Neurological (disease) | Mononeuritis multiplex, neuropathic pain | Low mortality | Very high morbidity — foot drop, chronic pain, disability |
| Renal (disease) | RPGN, CKD, ESRD | Significant (dialysis-dependent patients have ↑ mortality) | Dialysis dependence, CKD complications |
| Pulmonary (disease) | Chronic asthma, eosinophilic pneumonia, DAH | DAH can be fatal | Chronic dyspnoea, steroid dependence |
| GI (disease) | Mesenteric vasculitis, perforation | Perforation → peritonitis → high mortality | Chronic abdominal pain, malabsorption |
| Infections (treatment) | PJP, Strongyloides hyperinfection, HBV reactivation | #2 cause of death (after cardiac) | Recurrent infections, hospitalisations |
| Steroid toxicity (treatment) | Osteoporosis, DM, AVN, cataracts, Cushing's | ↑ cardiovascular mortality from metabolic syndrome | Major chronic morbidity burden |
| CYC toxicity (treatment) | Haemorrhagic cystitis, bladder CA, infertility | Bladder CA is a long-term cancer risk | Infertility in young patients |
High Yield Summary — Complications of EGPA
#1 cause of death = Cardiac (eosinophilic myocarditis → HF, arrhythmias, thromboembolism). This is unique to EGPA among the AAVs (GPA/MPA: pulmonary/renal death).
Three-stage cardiac damage: (1) Acute necrosis → (2) Thrombosis → (3) Fibrosis (restrictive CMP). Aggressive early immunosuppression aims to halt at Stage 1.
ANCA-negative phenotype → more cardiac complications; ANCA-positive → more renal/neurological vasculitic complications.
Neurological: Mononeuritis multiplex in 70–75% — vasa nervorum vasculitis → nerve infarction. Foot drop is classic. Recovery slow and often incomplete.
Renal: Pauci-immune GN (Type III RPGN) in ~45% — less common than GPA/MPA but can cause ESRD.
Treatment complications are the #2 cause of death: Infections (PJP, Strongyloides, HBV reactivation) from immunosuppression. Steroid toxicity causes enormous chronic morbidity.
Relapse rate: 25–50% at 5 years. Often triggered by steroid tapering. Each relapse → cumulative organ damage.
Thromboembolic risk: Eosinophils are prothrombotic (MBP inhibits thrombomodulin). Consider anticoagulation with intracardiac thrombus.
Active Recall - Complications of EGPA
References
[1] Senior notes: Maksim Medicine Notes.pdf (Rheumatology — EGPA three phases and eosinophilic infiltrative disease, p.331) [2] Senior notes: MBBS Final MB (Medicine) (Felix PY Lai).pdf (EGPA overview — cardiac involvement, ANCA phenotypes, pp.1767–1769) [3] Senior notes: Ryan Ho Rheumatology.pdf (AAV comparison table — clinical features, prognosis, pp.93–97) [5] Senior notes: Ryan Ho Respiratory.pdf (LTRA and Churg-Strauss unmasking, footnote 73, p.106) [6] Lecture slides: GC 053. Fingers turn white and blue.pdf (p.94 — EGPA vs GPA: major cause of death = cardiac) [18] Senior notes: Ryan Ho Cardiology.pdf (Myocarditis — management, prognosis, restrictive cardiomyopathy, pp.165–170)
High Yield Summary
Definition: EGPA (Churg-Strauss) = ANCA-associated necrotizing small-to-medium vessel vasculitis with eosinophil-rich granulomatous inflammation, asthma, and eosinophilia.
Epidemiology: Rare (0.5–6.8/million/year); age 20–40; slight male predominance.
Three Phases: (1) Allergic/Prodromal — asthma, allergic rhinitis, nasal polyps; (2) Eosinophilic — tissue infiltration (pneumonia, GI, myocarditis); (3) Vasculitic — mononeuritis multiplex, purpura, GN.
Key ANCA: p-ANCA (anti-MPO) positive in 40–50%. ANCA+ = more vasculitic (renal, nerves). ANCA− = more eosinophilic (cardiac, lung parenchyma).
Comparison Table (GPA vs EGPA vs MPA): EGPA is the only AAV with asthma and marked eosinophilia (> 10% WCC). Cardiac death is the major cause of mortality in EGPA (vs pulmonary/renal in GPA/MPA).
Histology: Eosinophilic necrotizing granuloma; pauci-immune on IF.
LTRA alert: Montelukast unmasks EGPA when steroids are tapered.
Cardinal clinical features: Asthma (> 90%), mononeuritis multiplex (75%), cardiac involvement (50%), eosinophilia (> 10%), palpable purpura, subcutaneous nodules.
High Yield Summary — DDx of EGPA
-
The unique triad of EGPA = Asthma + Eosinophilia (> 10% WCC) + Granulomatous vasculitis. No other condition produces all three together.
-
Among AAVs: EGPA is the only one with asthma and marked eosinophilia. GPA has ENT destruction + c-ANCA. MPA has no granulomas.
-
Key mimics to exclude: ABPA (Aspergillus serology, ↑↑ IgE, no vasculitis), HES (no asthma, no vasculitis, may have FIP1L1-PDGFRA), CEP (no vasculitis), parasitic eosinophilia (travel history, stool O&P).
-
For RPGN DDx: Use IF pattern — pauci-immune (Type III) = AAV; linear (Type I) = anti-GBM; granular (Type II) = immune complex. Complement levels help: ↓ C3/C4 = immune complex; normal = pauci-immune/anti-GBM.
-
LTRA unmasking: An asthmatic who develops systemic symptoms after steroid taper + LTRA initiation should be investigated for EGPA.
-
Cardiac cause of death in EGPA (vs pulmonary/renal in GPA/MPA) — this difference is frequently tested.
High Yield Summary — Diagnosis of EGPA
ACR 1990 Criteria: ≥ 4/6 of: asthma, eosinophilia > 10%, mononeuropathy/polyneuropathy, migratory pulmonary infiltrates, paranasal sinus abnormality, extravascular eosinophils on biopsy.
2022 ACR/EULAR Criteria: Weighted scoring; eosinophil count ≥ 1 × 10⁹/L carries +5 points; positive c-ANCA/anti-PR3 is a NEGATIVE criterion (−3) because it suggests GPA.
Key investigations: CBC D/C (eosinophilia > 10%), ESR/CRP (↑↑), ANCA (p-ANCA/anti-MPO in ~40–50%), complement (normal — rules out IC disease), urinalysis, CXR/HRCT, Echo/CMR, NCS/EMG, tissue biopsy.
Biopsy: Eosinophilic necrotising granuloma + necrotising vasculitis + pauci-immune IF. Skin biopsy is most accessible; sural nerve biopsy in mononeuritis multiplex; renal biopsy if GN suspected.
FFS: Cardiac, GI, renal insufficiency (Cr > 150), proteinuria > 1g/d, CNS. FFS ≥ 1 → add cyclophosphamide.
ANCA negative does NOT exclude EGPA — 50–60% are ANCA-negative.
High Yield Summary — Management of EGPA
Treatment framework: Induction → Maintenance, guided by Five-Factor Score (FFS).
FFS = 0 (non-severe): High-dose prednisolone alone (1 mg/kg/day, taper over 9–12 months).
FFS ≥ 1 (severe): IV pulse methylprednisolone (1 g/day × 3 days) + cyclophosphamide or rituximab.
Maintenance: Azathioprine / methotrexate / leflunomide + low-dose prednisolone for ≥ 18–24 months.
Biologic: Mepolizumab 300 mg SC Q4W — first-line for relapsing/refractory EGPA (blocks IL-5 → reduces eosinophil count). Benralizumab (anti-IL-5Rα) approved 2024.
EGPA vs GPA/MPA treatment: EGPA is the only AAV where steroids alone may suffice (FFS = 0); CYC is "±" not "+".
Key differences from GPA/MPA: CYC used selectively; cardiac management is critical; mepolizumab has specific role; asthma management is concurrent and lifelong.
Supportive care: PJP prophylaxis (co-trimoxazole), bone protection (calcium + vitamin D + bisphosphonate), HBV screening before rituximab, Strongyloides screening, gastroprotection.
Do NOT give NSAIDs in eosinophilic myocarditis — may worsen myocardial function.
Prognosis: 5-year survival 70–90%; cardiac death is the leading cause.
High Yield Summary — Complications of EGPA
#1 cause of death = Cardiac (eosinophilic myocarditis → HF, arrhythmias, thromboembolism). This is unique to EGPA among the AAVs (GPA/MPA: pulmonary/renal death).
Three-stage cardiac damage: (1) Acute necrosis → (2) Thrombosis → (3) Fibrosis (restrictive CMP). Aggressive early immunosuppression aims to halt at Stage 1.
ANCA-negative phenotype → more cardiac complications; ANCA-positive → more renal/neurological vasculitic complications.
Neurological: Mononeuritis multiplex in 70–75% — vasa nervorum vasculitis → nerve infarction. Foot drop is classic. Recovery slow and often incomplete.
Renal: Pauci-immune GN (Type III RPGN) in ~45% — less common than GPA/MPA but can cause ESRD.
Treatment complications are the #2 cause of death: Infections (PJP, Strongyloides, HBV reactivation) from immunosuppression. Steroid toxicity causes enormous chronic morbidity.
Relapse rate: 25–50% at 5 years. Often triggered by steroid tapering. Each relapse → cumulative organ damage.
Thromboembolic risk: Eosinophils are prothrombotic (MBP inhibits thrombomodulin). Consider anticoagulation with intracardiac thrombus.
Anti-GBM Disease
Anti-GBM disease is a small-vessel vasculitis caused by circulating autoantibodies directed against the alpha-3 chain of type IV collagen in glomerular and alveolar basement membranes, leading to rapidly progressive glomerulonephritis and, when pulmonary involvement occurs (Goodpasture syndrome), diffuse alveolar hemorrhage.
GPA Granulomatosis with Polyangiitis
Granulomatosis with polyangiitis (GPA) is a systemic necrotizing vasculitis of small- and medium-sized vessels characterized by granulomatous inflammation of the respiratory tract, necrotizing glomerulonephritis, and association with c-ANCA (anti-PR3) antibodies.